Zmiana funkcji termodynamicznych podczas adsorpcji. Termodynamika procesów adsorpcji. Projektowanie sprzętu procesów adsorpcji
Adsorpcja(z łac. ad - on, at i sorbeo - absorbuję), zmiana (zwykle - wzrost) stężenia substancji w pobliżu granicy faz („absorpcja na powierzchni”). Przyczyna adsorpcja- nienasycenie wiązań międzycząsteczkowych przy powierzchni, tj. istnienie adsorpcji pole siłowe. Ciało tworzące takie pole nazywa się adsorbentem, substancję, której cząsteczki mogą zostać zaadsorbowane, nazywa się adsorbatem, a substancję już zaadsorbowaną nazywa się adsorbatem. Proces odwrotny adsorpcja nazywa się desorpcją.
Charakter pola adsorpcyjnego jest inny. Jeśli adsorpcja jest powiązana z wiązaniami van der Waalsa, to wtedy adsorpcja zwany fizycznym. Jeżeli są to wiązania walencyjne, tj. adsorpcja przechodzi wraz z tworzeniem się powierzchni związki chemiczne, To adsorpcja zwany substancją chemiczną chemisorpcja. Ważne cechy chemisorpcja wykazuje: nieodwracalność, wysokie efekty termiczne (setki kJ/mol), charakter aktywowany. Istnieje wiele typów pośrednich adsorpcja pomiędzy fizycznym i chemicznym adsorpcja. Na przykład, adsorpcja spowodowane tworzeniem się wiązań wodorowych. Również możliwe różne rodzaje fizyczny adsorpcja. Występowanie dyspersyjnych sił przyciągania międzycząsteczkowego jest najczęstsze, ponieważ są one w przybliżeniu stałe dla adsorbentów o dowolnej powierzchni chemicznej (niespecyficzne adsorpcja). Fizyczny adsorpcja może być spowodowane siłami elektrostatycznymi (oddziaływanie jonów, dipoli lub kwadrupoli); w której adsorpcja zdeterminowany charakterem chemicznym cząsteczek środka adsorpcyjnego (tzw. specyficzny adsorpcja). Ważną rolę odgrywa także geometria interfejsu. jeśli powierzchnia jest płaska, to tak adsorpcja powierzchnia otwarta, w przypadku powierzchni lekko lub mocno zakrzywionej – ok adsorpcja w porach adsorbentu.
W teorii adsorpcja Rozróżnij statykę (układ adsorbent-adsorbat znajduje się w równowadze termodynamicznej) i kinetykę (nie ma równowagi).
Statyka adsorpcji
Termodynamika adsorpcji
.Podstawy termodynamiki adsorpcja zostały stworzone przez J. Gibbsa w latach 70-tych. 19 wiek Według Gibbsa w równowagowym układzie dwufazowym w pobliżu granicy faz następuje pewna zmiana lokalnych wartości wszystkich ekstensywnych właściwości (z wyjątkiem objętości). Fazy uważa się jednak za jednorodne aż do oddzielającej je powierzchni geometrycznej. Dlatego wartość jakiejkolwiek rozległej właściwości dla systemu jako całości nie jest równa sumie wartości tej właściwości w jednorodnych fazach i . Różnicę przypisuje się dwuwymiarowej fazie powierzchniowej związanej z powierzchnią oddzielającą. Ponieważ faza powierzchniowa nie ma wówczas grubości V0=+ i =-, gdzie V- tom.
Przedstawione reprezentacje pozwalają sprowadzić podstawowe równanie termodynamiczne do postaci:
gdzie G to energia swobodna Gibbsa, S to entropia, to napięcie międzyfazowe, s to obszar interfejsu, a n I- odpowiedni potencjał chemiczny i liczba moli I-ten komponent. Indeks wskazuje wartość danej właściwości w fazie powierzchniowej. Transformacja Legendre’a pozwala na modyfikację równania (1) dla warunków izotermicznych:
Ilość nazywa się Gibbs adsorpcja i jest oznaczony symbolem G (wyrażony w mol / cm2). Dla układu dwuskładnikowego:
Położenie powierzchni podziału można wybrać dowolnie. W szczególności wybór tego przepisu może spełniać warunek Г 1 =0. Taka powierzchnia nazywana jest równomolekularną. W tym celu wprowadza się oznaczenie Г 2 = Г 2 (1). Implikuje to główne równanie adsorpcji Gibbsa:
Jeżeli adsorbent jest całkowicie nierozpuszczalny w jednej z dwóch faz, =const, a przejście z równania (2) do równania (3) nie wymaga warunku Г 1 =0. Zatem Gibbs adsorpcja oznacza nadmiar tego składnika w rzeczywistym układzie dwufazowym w porównaniu z takim układem, w którym obie fazy byłyby ściśle jednorodne aż do powierzchni rozdzielającej. Oprócz nadwyżek Gibbsa adsorpcja, odgrywa ważną rolę w jej teorii adsorpcja, rozumiany jako pełna zawartość składnika I w kosmosie W, który wykazuje siły adsorpcji. Oznaczanie pełnej treści poprzez A i zakładając, że komponent I całkowicie nierozpuszczalny w jednej z faz masowych, mamy:
gdzie C I-stężenie I-ty składnik w fazie masowej. Dla małych S I:
Adsorpcja może wystąpić na dowolnej granicy faz pomiędzy dowolnymi dwiema fazami, w szczególności na granicy faz płyn-ciecz (ciecz-gaz, ciecz-ciecz) lub ciało stałe-ciecz (ciało stałe-gaz, ciało stałe-ciecz). W układach płyn-płyn α można zmierzyć jako funkcję i wyznaczyć eksperymentalnie Г 2 (1) za pomocą równania (3). W drugim przypadku, aby wyznaczyć G 2 (1), mierzy się dowolną metodą n I 0 , , oraz stężenia i-tego składnika w tych objętościach. Stąd obliczane jest G I(1) . Ta metoda nazywa się wolumetryczną (lub wolumetryczną). Metodą wagową (grawimetryczną) ilość określa się bezpośrednio I-ty komponent interfejsu.
Izoterma adsorpcji
.W równowagowym układzie adsorpcji parametrami określającymi równowagę są: ja ciśnienia cząstkowe R(lub z I) i temperatura T. Są one powiązane tzw. równaniem termicznym:
Na adsorpcja indywidualny adsorbent ( I=1) to równanie ma postać:
Trzy szczególne przypadki równanie termiczne(Gdy T. r Lub A- stałe) odgrywają w teorii szczególną rolę adsorpcja:
a=- równanie izotermy adsorpcja,
T=- równanie izobarowe adsorpcja,
R-- równanie izosteryczne adsorpcja.
Specyficzny typ funkcji i jest określony przez cechy rozważanego systemu. Jeśli na przykład jeden z nich jest znany z dowolnej wartości T= const, to oczywiście pozostałe dwa również stają się znane. W tym przypadku nie jest konieczna znajomość analitycznej postaci zależności. Można je podać empirycznie jako zbiór wartości a, r I T.
W teorii adsorpcja zwykle pytanie dotyczy postaci funkcji A=(p)r, tj. o równaniu izotermy adsorpcja. Problem ten związany jest z towarzyszącymi efektami termicznymi adsorpcja. Przy obliczaniu zmiany wartości głównych funkcji termodynamicznych w przypadku przejścia dn moli adsorpcji z fazy objętościowej do fazy powierzchniowej w układzie równowagi w temp p = const, możliwe są dwa przypadki: w pierwszym przypadku uwzględnia się jedynie przemianę adsorbatu w adsorbat, ponieważ adsorbent w adsorpcja termodynamicznie niezmienny i jego rolą jest służenie jako źródło pola adsorpcyjnego; w drugim brana jest pod uwagę również zmiana adsorbentu.
Ponieważ układ jest w równowadze, potencjały chemiczne adsorbatu i adsorbatu są takie same; Entropia adsorbatu z powodu zmniejszenia ruchliwości cząsteczek przy adsorpcja mniejsza niż entropia środka adsorpcyjnego. Dlatego w przypadku obojętnego adsorbentu entalpia jest zawsze ujemna, tj. adsorpcja egzotermiczny. Uwzględnienie zmiany entropii adsorbentu może zmienić ten wniosek. Przykładowo podczas sorpcji przez polimery substancji, w których polimer pęcznieje, entropia tego ostatniego (ze względu na wzrost ruchliwości makrocząsteczek) może wzrosnąć tak mocno, że adsorpcja staje się endotermiczny. Poniżej tylko egzotermia adsorpcja.
Rozróżnij ciepło całkowe, różniczkowe, izosteryczne i średnie adsorpcja. integralne ciepło Q równa utracie entalpii (at V= const - stała energia wewnętrzna) przy zmianie adsorpcja z 1 zanim 2(w konkretnym przypadku może to być 1 \u003d 0): Q \u003d - (H 2 - H 1). Wartość tę zwykle odnosi się do masy adsorbentu i wyraża w J/kg.
Ciepło różnicowe Q(J / mol) jest równe utracie entalpii dH kiedy to się zmienia A NA da. Wyraża się to postawą q = - (dH/da). To oczywiste
Ciepło izosteryczne q st przyjmuje się jako równe:
gdzie jest różnica między objętościami molowymi adsorbatu i adsorbatu. Można to wykazać  dla idealnego adsorbentu gazu:
dla idealnego adsorbentu gazu:
Znaczenie wstępu qsi ponieważ do jego oznaczenia nie są wymagane dane kalorymetryczne (takie jak Q I Q) i można to obliczyć za pomocą równania (9) z wyników pomiarów adsorpcja. Wprowadzamy również średnie ciepło Q(J/mol):
Ze wzrostem A parametr Q zawsze rośnie, a Q może się zmniejszyć, zwiększyć lub pozostać niezmieniona. Ze wzrostem A z niejednolitą powierzchnią adsorpcja występuje w coraz mniej aktywnych obszarach, co prowadzi do spadku Q. Jednak w tym przypadku zmniejszają się średnie odległości między zaadsorbowanymi cząsteczkami, w wyniku czego zwiększają się siły przyciągania między nimi, a Q wzrasta. O przebiegu zależności decyduje stosunek obu wymienionych efektów q=f(a). Bardzo duże A siły odpychające zaczynają dominować również w tym regionie. Q zawsze maleje wraz ze wzrostem A.
W przypadku bardzo małych pokryć powierzchni równanie izotermy adsorpcja ma postać równania Henry'ego:
gdzie K H jest współczynnikiem Henry'ego. Rzeczywiście, dla bardzo małych A Warstwa adsorpcyjna jest podobna do dwuwymiarowego gazu doskonałego, więc jej równanie stanu wygląda następująco: rt, gdzie jest ciśnieniem dwuwymiarowym, jest obszarem zajmowanym przez jeden mol substancji. Zatem biorąc pod uwagę, że =- i korzystając z równania (3) otrzymujemy równanie (12). Wymaga tego równanie Henry'ego Q był stały. W przypadku dużych wypełnień równanie to przestaje obowiązywać. Dlatego G. Freindlich (1906) zaproponował opis izoterm adsorpcja następujące równanie empiryczne (równanie Freundlicha):
Gdzie k I N- stałe. Równanie to jest często używane jako wzór interpolacyjny, chociaż w przypadku małych wartości R nie wchodzi w równanie (12) i jest bardzo duże R prowadzi do nieograniczonego wzrostu, co jest niezgodne z doświadczeniem A.
Rygorystyczna teoria izoterm adsorpcja stworzył I. Langmuir (1914-18). Teoria opiera się na poniższym. model: 1) powierzchnia adsorbentu to zbiór identycznych energetycznie centrów aktywnych, na których adsorbowane (zlokalizowane) są cząsteczki adsorbatu; 2) w jednym ośrodku zaadsorbowana jest tylko jedna cząsteczka; Na adsorpcja powstaje tylko jedna adsorpcja. warstwa (monowarstwa); 3) adsorpcja na to centrum nie ma wpływu adsorpcja na inne ośrodki, czyli interakcję. zaadsorbowane cząsteczki można pominąć.
Model Langmuira tzw. zlokalizowane monocząsteczkowe adsorpcja na jednolitej powierzchni. równanie izotermy adsorpcja być może pasujący do tego modelu. uzyskane przy pomocy metody (molekularno-kinetyczne, termodynamiczne, statystyczno-termodynamiczne). A więc adsorpcja równowagę można wyrazić w następujący sposób. schemat:
Bez cząsteczek. Adsorpcja w gazie + adsorpcja. centrum fazy złożonej (centrum zajęte)
Stężenie cząsteczek w gazie jest proporcjonalne do p, stężenia substancji wolnych. wartość centralna ( a t - a), Gdzie oraz T - całkowita liczba ośrodków, a-liczba zajętych ośrodków, stężenie adsorpcji. wartość kompleksów adsorpcja Zatem stała równowagi wynosi: K p \u003d p (a t - A)/ adsorpcja Stąd otrzymujemy równanie Langmuira:
Gdzie B-T. zwany adsorpcja współczynnik równy K p -1. W obszarze bardzo niskich ciśnień bp” 1 i a = (a m b) p, co odpowiada równaniu Henry'ego, w którym KH= jestem b. W obszarze bardzo wysokich ciśnień br 1 i aa t; w której adsorpcja nie jest już zależny od ciśnienia. Stała równowagi b-1 jest powiązany ze standardową wartością potencjału izobarycznego reakcji:
Model Langmuira wymaga, aby różnica. ciepło i entropia adsorpcja nie zależała od stopnia wypełnienia powierzchni.
równanie (14) jest wyrażeniem ścisłym odpowiadającym modelowi Langmuira, jednak w praktyce rzadko ma ono uzasadnienie, gdyż sam model jest wyidealizowany adsorpcja Doktryna adsorpcja z lat 20 XX wiek w środkach. Stopień zbudowano w oparciu o osłabienie lub wyeliminowanie jednego lub drugiego założenia Langmuira adsorpcja
Langmuir zaproponował już sposób opisywania adsorpcja na niejednorodnej powierzchni (tj. przy założeniu, że nie wszystkie środki są takie same). Łącząc identyczne ośrodki w grupy i zakładając, że równanie (14) ma zastosowanie do każdej grupy, możemy to założyć adsorpcja na całej powierzchni wyraża się sumą wyrazów równania (14):
Zakładając, że liczba adsorpcji centra mogą być opisana przez ciągłą funkcję rozkładu wartości wolnych. energia, Ya.B. Zeldovich otrzymał ze wzoru (16) dla funkcji wykładniczej równanie typu (13).
adsorpcja na niejednorodnych powierzchniach - duży rozdział teorii adsorpcja Jej główna zadanie-rozwiązanie równania całkowego:
Gdzie f(str) - tak zwana. empiryczny izoterma adsorpcja, -ta lub inna f-cja rozkładu liczby centrów na wartościach wolnych. energia,( b, p)- lokalna izoterma adsorpcja, co zwykle przyjmuje się jako izotermę Langmuira adsorpcja
Podjęto wiele prób odrzucenia drugiego założenia Langmuira. adsorpcja Na tej ścieżce teoria wielocząsteczkowa adsorpcja, zaproponowana przez S. Brunauera, P. Emmeta i E. Tellera (teoria BET). Teoria zakłada, że w temperaturze poniżej temperatury krytycznej każda cząsteczka zaadsorbowana w pierwszej warstwie (ciepło adsorpcji qi,) jest środkiem cząsteczek tworzących drugą warstwę i tak dalej. Zakłada się, że ciepło adsorpcja we wszystkich warstwach z wyjątkiem pierwszej jest równe ciepło kondensacji. Model ten prowadzi do równania:
Gdzie c = exp[(q 1 -)/RT]. równanie (18) we współrzędnych a, p/p s odpowiada krzywej S. We współrzędnych p/p,
izoterma adsorpcja zgodnie z równaniem (18) powinna być liniowa. Nachylenie tej prostej (zwykle w przedziale 0,05 p/p s 0,30) i wycięty przez nią odcinek na osi y dają wartości odpowiednio. Na I Z. Powszechne stosowanie teorii BET wynika z faktu, że jej autorzy tak naprawdę rozważają adsorpcja niezlokalizowane, zidentyfikuj stałą Na nie z liczbą odrębnych adsorbentów. centrach, ale z liczbą cząsteczek adsorbatu w pierwszej warstwie przy najbliższym upakowaniu (at R= p.s.). Dlatego wprowadzając ideę obszaru zajmowanego przez jedną cząsteczkę w tej warstwie, przyjmujemy:
Gdzie S- powierzchnię adsorbatu adsorpcja Z reguły mierzy się w tym celu izotermę adsorpcja azot i przyjmij to za jego cząsteczkę = 0,162 nm 2. Często wykonywane podobne obliczenia S według modelu Langmuira nie jest poprawne, ponieważ metoda ta oczywiście dotyczy tylko plików niezlokalizowanych adsorpcja
w teorii wielocząsteczkowej adsorpcja duży wkład wniósł J. de Boer, który eksperymentalnie wykazał, że zależność średniej liczby warstw (więcej niż pierwsza) na wszystkich powierzchniach jest bliska chemicznie. natura, od p/p s wyraża się krzywą uniwersalną (tzw. krzywą t). Umożliwia także oszacowanie pola powierzchni adsorbentów.
Podjęto próbę uwzględnienia w modelu Langmuira także interakcji. pomiędzy adsorberami. Cząsteczki. Tak więc T. Hill i J. de Boer uważają, że równanie stanu adsorpcji. warstwa jest dwuwymiarowym odpowiednikiem równania van der Waalsa, otrzymaliśmy co następuje. równanie izotermy adsorpcja:
gdzie= a/a t, a oraz b stałe równania van der Waalsa adsorpcja R. Fowlera i E. Guggenheima z uwzględnieniem interakcji. adsorber cząsteczek, wyprowadziłem równanie:
gdzie jest stałą związaną z parami interakcji cząsteczek.
Istnieje inny mechanizm prowadzący do dodatkowych adsorpcja adsorbentów poniżej wartości krytycznej. temperatura na porowatych adsorbentach przy stosunkowo wysokich wartościach p/p. Jest to kondensacja kapilarna. Jeżeli w porach tworzy się wklęsły menisk adsorbatu, rozpoczyna się w nim kondensacja p/p s Zgodnie z równaniem Kelvina:
gdzie jest napięcie powierzchniowe adsorbatu, V -jego objętość molowa, r-promień krzywizny menisku adsorpcja Kondensacja kapilarna prowadzi do gwałtownego wzrostu izotermy adsorpcja Często (choć nie zawsze) obserwuje się w tym przypadku tzw. adsorpcja histereza, tj. niedopasowanie adsorpcji. i desorbty. gałęzie izotermy. Z reguły wynika to z faktu, że kształt menisku przy adsorpcja i desorpcja nie są zgodne.
Kondensacja kapilarna służy do określenia wielkości porów adsorbentu adsorpcja Zgodnie z równaniem (22) dla każdej wartości p/p s obliczyć promień krzywizny menisku adsorpcja Z tego, biorąc pod uwagę grubość adsorpcji. warstwy (np. wzdłuż krzywej t), kształtu obszaru przejściowego od warstwy do menisku oraz zależności od krzywizny przy bardzo małych r , znaleźć rozmiar liniowy (efektywny promień r ef) porów wypełnionych w danym miejscu p/p. Objętość takich porów zależy od wzrostu adsorpcja w tym punkcie izotermy. Na podstawie uzyskanych danych budowana jest krzywa rozkładu objętości porów wzdłuż ich promieni. Metodę można zastosować przy ref 1,5 nm. Zwykle obliczenia przeprowadza się metodą desorpcji. gałęzie izotermy, ale bardziej rygorystyczne nowoczesne. teoria wymaga uwzględnienia obu gałęzi przy konstruowaniu krzywej.
Potencjalna teoria adsorpcji i teoria wypełniania objętościowego mikroporów. Model adsorpcja, zasadniczo różniący się od Langmuira, zaproponował w 1914 roku M. Polyaki. Zgodnie z tym modelem, w pobliżu powierzchni adsorbentu zachodzi potencjalna adsorpcja. pole siłowe zmniejsza się wraz z odległością od powierzchni. W rezultacie ciśnienie adsorbentu, które w dużej odległości od powierzchni wynosi p, w jego pobliżu wzrasta i w pewnej odległości osiąga wartość p s, przy której adsorbent ulega kondensacji. Objętość warstwy pomiędzy interfejsem a geomem. miejsce punktów gdzie p = p s , wypełniony cieczą, co przypisuje się normalnym wartościom fizycznym. właściwości cieczy luzem. Odwracalny izotermiczny praca i adsorpcja. siły, określone równaniem = RTlnp / p s, tzw. adsorpcja potencjał, a cała koncepcja jest teorią potencjału adsorpcja Dla danej objętości V adsorpcja warstwa jest potencjałem zależnym od temperatury (ze względu na niezależność sił dyspersji od temperatury). Ta niezmienność temperatury umożliwia ponowne obliczenia adsorpcja z jednego t-ry do drugiego, chociaż równania izotermy adsorpcja na podstawie podanej teorii nie można było tego wywnioskować. Model Polyani jest szeroko i z sukcesem stosowany przez wielu. autorów zawierała ona jednak dwa bardzo wrażliwe zapisy: 1) założenie o jak najlepszej adsorpcji. film ma normalne wartości fizyczne. właściwości cieczy w masie (założenie to nie zostało potwierdzone eksperymentami); 2) niezmienność temperaturowa funkcji =f(V), leżąca u podstaw teorii została w przybliżeniu potwierdzona eksperymentalnie tylko dla bardzo drobno porowatych adsorbentów.
Korzystając z teorii potencjału, M.M. Dubinin zaproponował i rozwinął teorię objętościowego wypełniania mikroporów (TOZM). Postulowano, że teoria ta ma zastosowanie wyłącznie do adsorbentów mikroporowatych. Cechą takich adsorbentów, w których wymiary liniowe porów wynoszą r1 nm, jest to, że cała objętość ich porów jest „wypełniona” adsorbentami. pole. Dlatego kiedy adsorpcja nie są wypełnione warstwami, ale objętościowo. Wartością w rozpatrywanym przypadku nie jest adsorpcja. potencjału i aż do znaku substancji chemicznej. potencjał adsorbatu, mierzony od poziomu substancji chemicznej. potencjał normalnej cieczy w tej samej temperaturze. Cały zestaw porów adsorbentu dzieli się na trzy klasy: mikropory ( R 0,6 nm), mezopory (0,6 nm-20 nm) i makropory ( R 20 nm). adsorpcja w mikroporach zachodzi według schematu TOZM, tj. objętościowo, w mezoporach – zgodnie z mechanizmem wypełniania warstwa po warstwie, zakończonego kondensacją kapilarną. Makropory podczas adsorpcji. równowaga nie odgrywa żadnej roli.
Wprowadzenie koncepcji rozkładu objętości porów f-tsii na wartości chemiczne. potencjał adsorbujący w nich, M.M. Dubinin i L. V. Radushkevich otrzymali równanie izotermy adsorpcji TOZM, które zwykle zapisuje się poniżej. formularz:

Gdzie n, E i 0 -parametry ( a 0 = a Na p = p.s.). Zależność od temperatury A 0:
gdzie= -(da0/dT); 0 0 = 0 w T \u003d T 0. Opcje P I mi praktycznie niezależny od temperatury. W większości przypadków P= 2. Tylko w przypadkach, gdy nagrzewa się początkowa adsorpcja bardzo duży n > 2. Aby przeliczyć izotermy adsorpcja z jednego środka adsorpcyjnego na drugi, przyjmuje się w przybliżeniu, że Mi 1 /E 2 P 1 /P= i że a 01 /a 02 V 1 /V 2 , gdzie P I- spadochron, Vi- objętość molowa adsorbentu adsorpcja
Każdy adsorbent mikroporowaty charakteryzuje się według TOZM dwoma parametrami: W- objętość mikroporów ( W 0 = = a 0 V 0) i E 0 -charakterystyka. energia; W0 i E0 odnoszą się do standardowego adsorbentu, zwykle benzenu.
Wykorzystując założenie, że w rzeczywistym adsorbencie występują pory o różnej wielkości i wprowadzając rozkład wartości E.S wariancja równa F. Stekli zaproponował uogólnienie równania (23), zwane równaniem Dubinina-Stöckliego:

Gdzie B0- stała związana z mi w równaniu (23) oraz y= ![]() . Ponieważ w adsorpcji technika naiba. upowszechniły się adsorbenty mikroporowate (węgle aktywne, zeolity, drobno porowate kserożele), TOZM ma zastosowanie nie tylko w fizyce i chemii. badań, ale także w obliczeniach inżynierskich.
. Ponieważ w adsorpcji technika naiba. upowszechniły się adsorbenty mikroporowate (węgle aktywne, zeolity, drobno porowate kserożele), TOZM ma zastosowanie nie tylko w fizyce i chemii. badań, ale także w obliczeniach inżynierskich.
Adsorpcja mieszanin gazów i cieczy. W praktyce zawsze mają do czynienia nie z pojedynczym adsorbentem, ale z mieszaniną gazów lub roztworów ciekłych. Dlatego konieczne jest uogólnienie teorii adsorpcja w przypadku adsorpcji wieloskładnikowej adsorpcja W zasadzie można zacząć od dowolnego modelu adsorpcja i rozszerzyć to na ten przypadek. Na adsorpcja mieszaninę gazów, osiąga się to nie tylko przez większe skomplikowanie równań, ale także poprzez wprowadzenie do nich dodatków. empiryczny parametry związane z interakcją lub z interakcją. heterogenicznych cząsteczek lub, bardziej ogólnie, z wpływem pewnego in-in na współczynnik. działalność innych. Dopiero model Langmuira pozwala otrzymać równanie izotermy adsorpcja mieszaniny bez parametrów nieujętych w równaniach adsorpcja indywidualne wejście-w. Aby to zrobić, wystarczy wziąć pod uwagę, że podczas adsorpcji k-tego składnika z mieszaniny I składniki stanowiące część adsorpcji. centra mogą być zajmowane przez inne cząsteczki. Dlatego:
Gdy adsorpcja roztworami ciekłymi, niezależnie od ich stężenia, wypełniana jest cała powierzchnia adsorbentu adsorpcja w konsekwencji adsorpcja cząsteczkom k-tego składnika towarzyszy przemieszczenie pewnej liczby cząsteczek pozostałych składników, tj. adsorpcja jest konkurencyjny.
Rozróżnij molekularne i jonowe adsorpcja rozwiązania. Pierwsze ma miejsce, gdy adsorpcja roztwory nieelektrolitów, drugi roztwór elektrolitów. Molekularny adsorpcja jest zwykle wyrażany wartości nadmiarowe. Konkurencyjny charakter adsorpcja powoduje wartość A m.b. zarówno pozytywne, jak i negatywne. wyrażający adsorpcja I-tego składnika jako f-tion jego ułamka molowego w roztworze x I-, mamy tego G I= O w x I= 0 i x I = 1 (możliwa zmiana objętość substancji podczas adsorpcji. warstwa jest pomijana). Dlatego izoterma adsorpcja ma jeden lub więcej skrajności.
równanie izotermy adsorpcja binarne roztwory nieelektrolitów, wiarygodnie uzasadnione termodynamicznie, mają postać:

gdzie indeks s oznacza adsorpcję. faza, - ( dn s 2 / dn s 1) pokazuje, ile moli drugiego składnika zostało przesuniętych przez jeden mol pierwszego, co stanowi różnicę między terminami (częściami standardowymi) substancji chemicznej. potencjał zależny tylko od temperatury.
Główny problem wykorzystania tego i szeregu innych równań izoterm adsorpcja-wyznaczenie zależności współczynnika. aktywność składników w adsorpcji. warstwę ze swojego składu adsorpcja Najważniejsze pytanie we wniosku adsorpcja do separacji lub oczyszczania substancji - dobór selektywnego adsorbentu w stosunku do tego składowego roztworu adsorpcja
joński adsorpcja z reguły nie jest równoważny adsorpcja Preimy są adsorbowane na powierzchni z roztworu elektrolitu. kationy lub aniony. Dzięki elektrycznemu Powstają siły (kulombowskie) działające na powierzchnię podwójna warstwa elektryczna.
Jeśli adsorbent zawiera jony lub func. grupy zdolne do jonizacji w danym rozpuszczalniku, wówczas następuje wymiana jonowa pomiędzy adsorbentem a roztworem elektrolitu. Adsorbent w tym przypadku nazywa się. wymiennik jonowy.
Kinetyka adsorpcji
adsorpcja, jak każdy rzeczywisty proces, zachodzi w czasie. A więc pełna teoria adsorpcja powinien zawierać sekcję dotyczącą kinetyki adsorpcja akt elementarny adsorpcja przeprowadza się niemal natychmiastowo (z wyjątkiem chemisorpcji). A więc zależności czasowe adsorpcja są zdefiniowane w części głównej mechanizm dyfuzyjny, czyli dostarczanie adsorpcji do miejsca adsorpcja Jeśli adsorpcja na otwartej powierzchni nie jest natychmiastowy, proces taki zachodzi w zewnętrznym obszarze dyfuzji; podczas gdy prawa dyfuzji nie są specyficzne dla adsorpcja W przypadku adsorbentów porowatych oprócz ext. dyfuzji, ważną rolę zaczyna odgrywać vnutr. dyfuzja, tj. transfer adsorbentu w porach adsorbentu przy obecności w nich gradientu stężeń. Mechanizm takiego przenoszenia może zależeć od stężenia adsorbatu i wielkości porów.
Wyróżnia się dyfuzję molekularną, Knudsena i powierzchniową (Volmera). Dyfuzję molekularną przeprowadza się, jeśli długość jest wolna. zakres cząsteczek w porach jest mniejszy niż rozmiar porów, długość Knudsena ma miejsce, jeśli długość ta przekracza rozmiar porów. Podczas dyfuzji powierzchniowej cząsteczki przemieszczają się po powierzchni adsorbentu bez przejścia do fazy objętościowej. Jednak wartości współczynnika dyfuzje nie są takie same dla różnych mechanizmów dyfuzji. W wielu przypadkach nie jest możliwe ustalenie eksperymentalnie dokładnie, w jaki sposób zachodzi dyfuzja, a co za tym idzie tzw. efektywny współczynnik. dyfuzja, opisująca proces jako całość.
Główny eksperymentalny materiał o kinetyce adsorpcja służy tzw. kinetyczny krzywa, tj. f-tion \u003d a / a równy \u003d f(t) gdzie jest względne adsorpcja równy stosunkowi bieżącej wartości adsorpcji A Do A równa jego wartości w danym momencie T. Interpretacja kinetyki W najprostszym przypadku przyjmuje się, że ziarno adsorbentu ma całkowicie jednolitą strukturę porowatą pod względem objętości (model ten nazywa się quasi-homogenicznym). Oznacza. udoskonalenie modelu quasi-jednorodnego - poglądu, że każde ziarno zawiera obszary o większych i drobniejszych porach. Dyfuzja w takim ziarnie opisana jest przez dwa dec. współczynniki.
W przypadku powierzchni otwartej, korzystając z modelu Langmuira, łatwo jest uzyskać kinetykę. równanie adsorpcja Szybkość zbliżania się do równowagi jest różnicą prędkości adsorpcja i desorpcja. Zakładając, jak zwykle w kinetyce, że szybkości procesów są proporcjonalne do stężeń reagujących substancji, mamy:
gdzie k ad i k dec są odpowiednio stałymi szybkości. adsorpcja i desorpcja. Zakłada się, że ciśnienie w fazie gazowej jest stałe. Całkując to równanie z T= 0 do dowolnej wartości T otrzymujemy:
Zatem dla f mamy:= równe. Więc w końcu mamy:
gdzie k = k reklam + k dec.
Wpływ temperatury na prędkość adsorpcja wyrażone równaniem podobnym do równania Arrheniusa adsorpcja Wraz ze wzrostem temperatury k reklam wzrasta wykładniczo. Ponieważ dyfuzja w porach adsorbentu wiąże się z przezwyciężeniem aktywacji. bariery, zależności temperaturowe kad i kdes nie są takie same.
Znajomość szybkości dyfuzji jest ważna nie tylko dla teorii adsorpcja, ale także do obliczenia balu. adsorpcja procesy. W tym przypadku zwykle nie zajmują się pojedynczymi ziarnami adsorbentu, ale ich warstwami. Kinetyka procesu w warstwie wyraża się bardzo złożonymi zależnościami. W każdym punkcie warstwy w danym momencie wartość adsorpcja zależy nie tylko od postaci równania izotermy adsorpcja i prawa kinetyki procesu, ale także aero- czy hydrodynamiczne. warunki przepływu gazu lub cieczy wokół ziaren. Nazywa się kinetyką procesu w warstwie adsorbentu, w przeciwieństwie do kinetyki w pojedynczym ziarnie. dynamika adsorpcja, ogólny schemat rozwiązywanie problemów, które polegają na: kompilowaniu układu różniczkowego. równania w pochodnych cząstkowych z uwzględnieniem charakterystyki warstwy, izotermy adsorpcja, właściwości dyfuzyjne (współczynnik dyfuzji, rodzaje przenoszenia masy wzdłuż warstwy i wewnątrz ziaren), aero- i hydrodynamiczne. cechy przepływu adsorpcja Ustawiono warunki początkowe i brzegowe. Rozwiązanie tego układu równań prowadzi w zasadzie do wartości wielkości adsorpcja w danym momencie w danym punkcie warstwy. Z reguły analityczny rozwiązanie można uzyskać tylko dla najprostszych przypadków, dlatego taki problem rozwiązuje się numerycznie za pomocą komputera.
W eksperymentalnym badaniu dynamiki adsorpcja przez warstwę adsorbentu przepuszcza się strumień gazu lub cieczy o określonej charakterystyce i bada się skład wypływającego strumienia w funkcji czasu. Pojawienie się wchłoniętej substancji za warstwą tzw. przełom, a czas przełomu – czas działania ochronnego. Zależność stężenia tego składnika za warstwą od czasu tzw. krzywa wyjściowa. Krzywe te służą jako główne eksperymentalny materiał, który umożliwia ocenę wzorców dynamiki adsorpcja
Projektowanie sprzętu procesów adsorpcji
Istnieje wiele technologii. techniki adsorpcji. procesy. Powszechna cykliczność. Instalacje (okresowe) ze stałym złożem adsorbentu, osn. którego węzeł jest jeden lub kilka. adsorbery wykonane w formie pustych kolumn wypełnionych granulowanym adsorbentem. Strumień gazu (lub cieczy) zawierający zaadsorbowane składniki przepuszcza się przez złoże adsorbentu aż do przebicia adsorpcja Następnie następuje regeneracja adsorbentu w adsorberze, a strumień gazu kierowany jest do innego adsorbera. Regeneracja adsorbentu składa się z kilku etapów, z których głównym jest desorpcja, czyli tzw. uwolnienie wcześniej zaabsorbowanej materii z adsorbentu adsorpcja Desorpcję przeprowadza się poprzez ogrzewanie, rozprężanie fazy gazowej, wypieranie (np. pary świeżej) lub kombinację tych metod. Ponieważ czasy adsorpcja i regeneracja nie pokrywają się, należy dobrać taką liczbę jednocześnie pracujących i regenerowanych adsorberów, aby cały proces przebiegał w sposób ciągły.
Według tech. i ekonomiczne względy regeneracja nie jest zakończona adsorpcja Zatem pojemność robocza adsorbentu jest równa różnicy pomiędzy maksymalną osiągalną w danych warunkach adsorpcja oraz ilość adsorbatu pozostałego w adsorbencie po regeneracji. W rezultacie izotermy adsorpcja odpowiadająca procesowi w adsorberze nie powinna być zbyt stroma.
W opisywanym schemacie możliwe są dwie opcje: 1) docelowy produkt jest prawie całkowicie adsorbowany ze strumienia gazu, a następnie zostaje zawarty w desorbacie, skąd jest w ten czy inny sposób ekstrahowany; 2) docelowy produkt jest adsorbowany gorzej niż inne składniki mieszaniny gazowej i wówczas zostaje zawarty w wypływającym strumieniu gazu. Zgodnie z pierwszą opcją działają na przykład jednostki rekuperacji w fabrykach wiskozy, wychwytując ze gazów spalinowych i zawracając CS 2 do obiegu. Wydajność takich instalacji sięga setek tysięcy m 3 oczyszczonego gazu na godzinę; adsorbentowy węgiel aktywny z niezbyt drobnymi mikroporami, tj. węgiel, w którym stała mi według TOZM (patrz wyżej) wynosi 20-25 kJ / mol. Ta wartość mi 0 odpowiada niezbyt stromej izotermie, która zapewnia dobre warunki regeneracji. Takie węgle nazywane są powrót do zdrowia. Desorpcję przeprowadza się za pomocą pary świeżej. Aby oszczędzać energię, zimne i gorące gazy przepuszczane są przez wymienniki ciepła.
Bardzo ważne jest na przykład suszenie gazów i cieczy gazy ropopochodne przed obróbką lub naturalne. gazy przed transportem; adsorbenty w postaci żelu krzemionkowego lub zeolity. Desorpcję przeprowadza się przez ogrzewanie. Ponieważ desorpcja zeolitu wiąże się z wysokimi kosztami energii, stosuje się adsorbent kombinowany: główny. masa wilgoci zostaje wchłonięta przez łatwo regenerowany żel krzemionkowy, a głębokie dosuszenie przez zeolit.
Podczas regeneracji termicznej pełny cykl obejmuje adsorpcja, ogrzewanie adsorbentu, jego desorpcja i chłodzenie. Duża liczba Etapy determinują niską i wysoką energochłonność procesu adsorpcja Dlatego tzw. instalacje krótkocykliczne, których cały cykl trwa kilka. minuty. W nich gaz jest dostarczany do adsorbera pod średnią. ciśnienie, które następnie zostaje uwolnione i następuje desorpcja. Cały proces jest niemal izotermiczny (odchylenie od izotermii spowodowane jest jedynie wydzieleniem ciepła adsorpcja i absorpcja ciepła podczas desorpcji). Etapy cyklu: adsorpcja, rozprężenie, desorpcja, wzrost ciśnienia. Przykładem jest instalacja wykorzystująca zeolit do produkcji powietrza wzbogaconego w tlen.
W instalacjach z ruchomą warstwą adsorbentu (w tzw. hipersorberach) ten ostatni pod wpływem grawitacji powoli opada, jest usuwany z dna. części adsorbera i wchodzi do tzw. podnośnik powietrzny, który jest pionową rurą równoległą do adsorpcji. kolumna. Strumień powietrza przepływa przez tę rurę od dołu do góry, co unosi ziarna adsorbentu do góry. część kolumny. Strumień przetworzonego gazu wpływa do środkowej części adsorbera i przemieszcza się w górę w kierunku przeciwnym do adsorbentu. Na górze kolumny znajduje się ciągła adsorpcja, na dole - regeneracja adsorbentu (patrz też czyszczenie adsorpcyjne).
W instalacjach z fluidalnym („wrzącym”) złożem adsorbentu przepływ gazu wchodzący do adsorbera od dołu powoduje przejście adsorbentu w zawiesinę. To znacznie zwiększa efektywność przenoszenia masy pomiędzy adsorbentem a gazem i skraca czas trwania adsorpcja i desorpcja. Takie instalacje mają wysoką wydajność. Ich szerokie rozpowszechnienie utrudniają wysokie wymagania dotyczące futra. wytrzymałość ziaren adsorbentu (niewystarczająca wytrzymałość powoduje znaczne straty adsorbentu na skutek jego ścierania i wyrywania z aparatu).
Główny wymagania dotyczące adsorbentów: adsorbent duży. pojemność, tj. powinny to być ciała rozproszone, z dużym rytmem. powierzchnia lub o dużej objętości porów; chemia charakter powierzchni musi zapewniać skuteczność adsorpcja dane wprowadzane w tych warunkach; chemia i termiczne. trwałość, regenerowalność, dostępność. maks. powszechne stały się węgle aktywne, kserożele niektórych tlenków (żele krzemionkowe, żele tlenku glinu itp.), Zeolity; z nieporowatych adsorbentów-tech. węgiel (sadza) i silnie zdyspergowany SiO 2 (aerosil, „biała sadza”).
Obszary zastosowań technologii adsorpcyjnej
O zjawisku adsorpcja założona przez wielu sposoby oczyszczania powietrza ze szkodliwych zanieczyszczeń (patrz. oczyszczanie gazu), woda (patrz Uzdatnianie wody), a także syropy cukrowe do produkcji cukru, soki owocowe i inne płyny w żywności. prom-sti, odpadowe oleje smarowe. Usuwanie wilgoci jako szkodliwego zanieczyszczenia z gazów i cieczy za pomocą stałych adsorbentów jest jedną z ważnych gałęzi adsorpcji. techniki (patrz także suszenie gazowe).
O adsorpcji. procesy opierają się na dokładnym rozdzieleniu mieszanin substancji i wyodrębnieniu niektórych składników ze złożonych mieszanin. Przykładami są separacja izomerów alkanów w celu otrzymania normalnych węglowodorów do produkcji środków powierzchniowo czynnych, separacja olejów przy produkcji paliw silnikowych. Dla mieszaniny gazów adsorpcja metody separacji stosowane są w celu uzyskania powietrza wzbogaconego w tlen (aż do prawie czystego O 2); w wielu przypadkach metody te z powodzeniem konkurują z destylacją (por. separacja powietrza).
Szybko rozwijająca się dziedzina zastosowań adsorbentów. technologia-medycyna, gdzie służy wydobyciu szkodliwe substancje z krwi (metoda hemosorpcji) itp. fiziol. płyny. Wysokie wymagania dotyczące sterylności stawiają bardzo trudne zadanie doboru odpowiednich adsorbentów. Należą do nich specjalnie przygotowane węgle aktywne.
Oświetlony.: Brunauer S., Adsorpcja gazów i par, przeł. z języka angielskiego, t. 1, M., 1948; de Boer Ya, Dynamiczna natura adsorpcji, przeł. z języka angielskiego, M., 1962; Adsorpcja i porowatość, wyd. M. M. Dubinina [i in.], M., 1976; Keliev N.V., Podstawy technologii adsorpcji, wyd. 2, M., 1984; Young D.M., Crowell A.D., Fizyczna adsorpcja gazów, L., 1962. M.M. Dubinin, V.V. Serpińskiego.
Wybierz pierwszą literę w tytule artykułu:
Adsorpcja zachodzi na granicy faz. Dlatego zasadne jest rozważenie termodynamicznego opisu zjawisk powierzchniowych jako szczególny przypadek termodynamika układów heterogenicznych.
Ryż. 3.4. Adsorpcja Gibbsa: 1- dwufazowy układ odniesienia, 2- rzeczywisty układ dwufazowy z niejednorodnym obszarem
Wykorzystuje termodynamikę układów heterogenicznych zasada addytywności, czyli następująco: wszystkie rozległe właściwości układu heterogenicznego są równe sumie odpowiednich rozległych właściwości, jakie miałyby fazy przed ich zetknięciem. Oznaczmy fazy przez α i β (ryc. 4). Następnie dla układu idealnego, w którym właściwości faz w pobliżu granicy faz pokrywają się z ich właściwościami masowymi, dla energii wewnętrznej U, objętości V, masy (liczby moli) n, entropii S po ustaleniu się równowagi w układzie heterogenicznym, relacje są ważne:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
Zakłada się, że temperatura i ciśnienie w obu fazach są takie same.
W przypadku rzeczywistych układów heterogenicznych obszar przejściowy na styku dwóch faz wnosi dodatkowy wkład w rozległe właściwości układu. Jeżeli występują zjawiska powierzchniowe, należy wziąć pod uwagę różnicę między ekstensywnymi właściwościami rzeczywistego układu heterogenicznego a ekstensywnymi właściwościami układu modelowego, w którym nie występują zjawiska powierzchniowe. Taki system nazywa się systemem porównawczym. System porównawczy ma te same parametry intensywne (T, P, C i…) i taką samą objętość V jak system rzeczywisty (rys. 4).
Z termodynamicznego punktu widzenia wartość adsorpcji G jest rozumiana jako nadmiarowa ilość substancji n s, wyrażona w molach lub gramach, jaką ma rzeczywisty układ heterogeniczny w porównaniu z układem odniesienia, w odniesieniu do pola powierzchni fazy separacji lub do powierzchni adsorbentu A. Zakłada się, że układ porównawczy ma te same parametry intensywne (T, P, C i) i tę samą objętość (V = V α + V β) co układ rzeczywisty (ryc. 4).
G \u003d (n - n α - n β) / A \u003d n s / A 3.11
Nadmiarowe funkcje termodynamiczne obszaru przejściowego układu rzeczywistego (oznaczone indeksem s) można zapisać jako
U s = U - U α - U β , n s = n - n α - n β , S s \u003d S - S α - S β itp.
Eksperymentalne pomiary adsorpcji zawsze dają adsorpcję dokładnie jako nadmiar składnika prawdziwy system w porównaniu z wybranym systemem porównawczym. Przykładowo, podczas adsorbowania gazu na adsorbencie stałym lub podczas adsorbowania składników na fazie stałej, aby znaleźć wartości adsorpcji, wyznacza się zmianę początkowych stężeń adsorbatu po zetknięciu faz α i β
n ja s = V(C ja o - C ja),
Gdzie C i o– początkowe stężenie i-tego składnika, C ja jest stężeniem i-tego składnika po ustaleniu się równowagi pomiędzy sąsiadującymi fazami. Zakłada się, że objętość V nie zmienia. Jednak koncentracja I-ty składnik C ja, otrzymany doświadczalnie, określa się w objętości V' nad interfejsem bez uwzględnienia objętości niejednorodnego obszaru warstwy przejściowej Va na granicy faz, gdzie występuje stężenie C i α. Zatem ze względu na istnienie niejednorodnego obszaru w układzie rzeczywistym całkowitą objętość układu można przedstawić jako V = V' + Va. Cała ilość I-ty składnik C i o rozdzielone pomiędzy tymi dwoma tomami:
V do ja o = V' do ja + V α do ja α ,
i liczbę moli składnika I, zaadsorbowany na granicy faz, będzie równy
n ja s = (V’C i + V α C i α) – (V’ + V α)C i = V α (C i α – C i) 3.12
Te. adsorpcja określona doświadczalnie to nadmiar i-tego składnika w objętości V α w porównaniu z ilością tego składnika w tej samej objętości oddalonej od granicy faz. Ten typ adsorpcji nazywa się adsorpcją Gibbsa. .
V α C i α zwany pełna treść I- składnik warstwy adsorpcyjnej. W obszarze bardzo niskich stężeń C ja w objętości V' poprawka V α C ja równanie (3.2) można pominąć i uwzględnić zmierzoną wartość V α C i α pełna treść I- składnik warstwy adsorpcyjnej, na przykład podczas adsorpcji gazu na stałym adsorbencie pod niskim ciśnieniem.
Bieżąca strona: 6 (książka ma łącznie 19 stron) [dostępny fragment lektury: 13 stron]
Czcionka:
100% +
34. Charakter sił adsorpcji
Oddziaływanie cząsteczek adsorbentu z powierzchnią adsorbentu na tzw. Adsorpcja fizyczna może mieć różne przyczyny. Wówczas potencjał decydujący o oddziaływaniu jednej cząsteczki adsorbentu z jednym atomem adsorbentu niepolarnego można wyrazić następująco:
θ = −Kr 6 +br 12 ,
gdzie r jest odległością między środkami cząstek; C jest stałą przyciągania dyspersji; B jest stałą charakteryzującą energię sił odpychających.
Jest rzeczą oczywistą, że na stosunkowo dużych odległościach powinny przeważać siły przyciągające, a na małych – siły odpychające. Ponadto w pewnych odległościach siły te muszą być równe, co będzie odpowiadać minimum Darmowa energia. Należy jednak zauważyć, że podczas adsorpcji siły dyspersji działają jednocześnie pomiędzy każdą cząstką niepolarną.
Ponieważ energia oddziaływania cząstek może gwałtownie spadać wraz z odległością, wystarczy wykonać sumowanie na najbliższych atomach adsorbentu, aby określić potencjał sił adsorpcji. Ważne jest, aby w przypadku adsorpcji złożonych cząsteczek niepolarnych energię potencjalną można było w przybliżeniu obliczyć jako sumę wszystkich potencjalnych energii adsorpcji jednostek cząsteczki.
Jeżeli adsorbent składa się z jonów, wówczas działanie znanych już sił dyspersyjnych można uzupełnić działaniem sił indukcyjnych przyciągania dipoli, które indukowane są w cząsteczkach adsorbentu przez pole elektryczne, które z kolei powstaje przez jony sieci adsorbentu.
Przy takim oddziaływaniu udział sił indukcyjnych w oddziaływaniu adsorpcyjnym może być proporcjonalny do polaryzowalności cząsteczki adsorpcyjnej i kwadratu natężenia pola na tej powierzchni adsorbentu.
Jeżeli natomiast na adsorbencie polarnym adsorbowane są cząsteczki polarnego adsorbentu, to dipole w tym przypadku polaryzują atomy adsorbentu, czyli tak jakby indukowały w nich momenty elektryczne. Ze względu na ten wpływ do oddziaływania dyspersyjnego dodaje się oddziaływanie indukcyjne.
Samo oddziaływanie indukcyjne jest zwykle małe i w zależności od dipola cząsteczki adsorpcyjnej i polaryzowalności adsorbentu może osiągać duże wartości. W przypadku, gdy cząsteczki są adsorbowane na adsorbencie, który ma na powierzchni jony lub dipole, dochodzi do tzw. oddziaływanie jonów lub dipoli adsorbentu z polem elektrostatycznym samego adsorbentu.
W tym przypadku cząsteczki adsorpcyjne mogą nawet zorientować się w polu adsorbentu i zachodzi orientacyjne oddziaływanie kulombowskie. Zwykle zdarza się, że energie oddziaływań indukcyjnych i orientacyjnych są mniejsze od energii oddziaływań dyspersyjnych, dlatego przyjmuje się, że o energii przyciągania międzycząsteczkowego decyduje energia przyciągania dyspersyjnego.
Przyczyną adsorpcji może być również utworzenie wiązania wodorowego. Wiązanie tego typu może powstać podczas adsorpcji na adsorbentach zawierających na powierzchni grupy hydroksylowe cząsteczek, takich jak woda, alkohole, amoniak i aminy. Kiedy tworzy się wiązanie wodorowe, energia oddziaływania adsorbentu z adsorbentem może być dość duża, a ciepło wydzielające się podczas takiej adsorpcji jest znacznie większe niż ciepło adsorpcji substancji o podobnym kształcie i wielkości cząsteczek, ale nie tworzą wiązania wodorowego.
Należy zaznaczyć, że znając opis termodynamiczny warstwy powierzchniowej na granicy „adsorbent – adsorbent”, jej strukturę, charakter różnego rodzaju sił, dynamikę procesu, można przystąpić do badań bardziej złożonych procesy adsorpcji.
35. Adsorpcja jako samoistne stężenie na granicy faz substancji zmniejszających napięcie międzyfazowe
Surfaktanty dzielą się na dwie duże grupy: aktywne i nieaktywne Substancje.
Środki powierzchniowo czynne mogą gromadzić się w warstwie powierzchniowej i w tym przypadku następuje dodatnia adsorpcja. G > 0.
Tego rodzaju substancje muszą mieć napięcie powierzchniowe, które z kolei musi być mniejsze od napięcia powierzchniowego rozpuszczalnika, w przeciwnym razie gromadzenie się substancji w warstwie powierzchniowej byłoby niekorzystne i musi charakteryzować się stosunkowo niską rozpuszczalnością. Przy wystarczająco dobrej rozpuszczalności cząsteczki środka powierzchniowo czynnego mają tendencję do opuszczania powierzchni głęboko w roztworze. Dlatego środki powierzchniowo czynne będą preferencyjnie wypychane z większości cieczy na powierzchnię.
Ale wraz z gromadzeniem się substancji na granicy roztworu w cząsteczkach tych substancji, które słabo ze sobą oddziałują, oddziaływanie międzycząsteczkowe w warstwie powierzchniowej zmniejszy się, a napięcie powierzchniowe spadnie.
Środki powierzchniowo czynne w stosunku do warstwy wodnej występuje wiele rodzajów związków organicznych, kwasy tłuszczowe z odpowiednio dużym rodnikiem węglowodorowym, sole tych kwasów (mydła), kwasy sulfonowe i ich sole, a także Różne rodzaje alkohole i aminy. cecha charakterystyczna większość cząsteczek to ich difilowość: cząsteczka składa się z dwóch części grupy polarnej i niepolarnego rodnika węglowodorowego. Posiadanie znacznego momentu dipolowego i dobrze uwodniającej grupy polarnej może określić powinowactwo środka powierzchniowo czynnego do środowiska wodnego. Jednak przyczyną obniżającą rozpuszczalność tych związków jest rodnik węglowodorowy.
Powierzchniowo nieaktywne środki powierzchniowo czynne- tego typu substancje, mające tendencję do opuszczania powierzchni cieczy do jej objętości, w efekcie tzw. ujemna adsorpcja G < 0. Поверностно-инактивные вещества также обладают значительным поверхностным натяжением, значительно большим, чем натяжение у растворителя (иначе эти вещества способны самопроизвольно накапливаться в поверхностном слое), также обладают высокой растворимостью, что способствует их стремлению уйти с поверхности жидкости в объем. Взаимодействие между молекулами поверхностно-инактивного вещества и растворителя всегда больше, чем взаимодействие между самими молекулами растворителя, поэтому они и стремятся перейти в объем раствора. Substancje nieaktywne powierzchniowo w stosunku do wody występuje wiele elektrolitów nieorganicznych: kwasy, zasady, sole. Cząsteczki substancji powierzchniowo nieaktywnych nie mają części hydrofobowej i mogą rozkładać się w wodzie na silnie nawilżające jony.
Przykłady Substancje nieaktywne powierzchniowo to także niektóre związki organiczne, w których niepolarna część cząsteczki jest nieobecna lub jest bardzo mała. Substancje te obejmują kwas mrówkowy i aminooctowy.
W rozpuszczalnikach niewodnych elektrolity nieorganiczne mogą również zwiększać napięcie powierzchniowe, a to zależy od rozpuszczalnika.
Na przykład, po wprowadzeniu jodku sodu do metanolu napięcie powierzchniowe znacznie wzrasta, w przypadku etanolu napięcie powierzchniowe jest około 2 razy większe. Aktywność powierzchniowa substancji może zależeć nie tylko od charakteru substancji, ale także od właściwości rozpuszczalnika. Jeśli jakikolwiek rozpuszczalnik ma wysokie napięcie powierzchniowe, wówczas ta substancja rozpuszczona może wykazywać znaczną aktywność powierzchniową.
36. Teorie adsorpcji
Rozważmy najpopularniejsze teorie adsorpcji, które opisują poszczególne typy adsorpcji na granicy faz ciało stałe-gaz lub ciało stałe-roztwór.
Teoria adsorpcji jednocząsteczkowej I. Langmuira.
1. Adsorpcja ma charakter lokalny i jest spowodowana siłami zbliżonymi do chemicznych.
2. Adsorpcja zachodzi tylko na centrach aktywnych - występach lub zagłębieniach na powierzchni adsorbentu, charakteryzujących się obecnością wolnych wartościowości. Ośrodki aktywne uważane są za niezależne i identyczne.
3. Każde centrum aktywne może oddziaływać tylko z jedną cząsteczką adsorbatu; na powierzchni może utworzyć się tylko jedna warstwa zaadsorbowanych cząsteczek.
4. Proces adsorpcji jest odwracalny i równowagowy; zaadsorbowana cząsteczka jest zatrzymywana przez centrum aktywne przez pewien czas, po czym ulega desorpcji; Po pewnym czasie ustala się dynamiczna równowaga.
Maksymalna możliwa wartość adsorpcji G o osiąga się pod warunkiem, że wszystkie centra aktywne zostaną zajęte przez cząsteczki adsorbatu. Równanie izotermy adsorpcji monocząsteczkowej odnoszące się do wartości adsorpcji G ze stężeniem adsorbatu Z, wygląda jak:
Gdzie B- stała dla danej pary wartości „adsorbent - adsorbat” (stosunek stałych szybkości desorpcji i adsorpcji), liczbowo równa stężeniu adsorbatu, przy którym zajęta jest połowa centrów aktywnych.

Wykres izotermy adsorpcji Langmuira pokazano na rysunku 2. Stała B definiujemy graficznie, rysując styczną do izotermy adsorpcji w tym punkcie Z= 0. Opisując proces adsorpcji gazów w równaniu, stężenie można zastąpić proporcjonalną wartością ciśnienia cząstkowego. Teoria adsorpcji monocząsteczkowej I. Langmuira ma zastosowanie do opisu procesów adsorpcji gazów i substancji rozpuszczonych przy niskich ciśnieniach (stężeniach) adsorbatu.
Teoria adsorpcji wielocząsteczkowej Polanyi'ego opisuje izotermy adsorpcji w kształcie litery S, których kształt wskazuje na możliwe oddziaływanie zaadsorbowanych cząsteczek z adsorbatem.
1. Adsorpcja jest spowodowana siłami fizycznymi.
2. Powierzchnia adsorbentu jest jednorodna, nie ma tam centrów aktywnych; siły adsorpcji tworzą ciągłe pole siłowe w pobliżu powierzchni adsorbentu.
3. Siły adsorpcji działają w odległości większej niż wielkość cząsteczki adsorbatu, tzn. w pobliżu powierzchni adsorbentu, która podczas adsorpcji jest wypełniona cząsteczkami adsorbatu, występuje pewna objętość adsorpcji.
4. Przyciąganie cząsteczki adsorbatu przez powierzchnię adsorbentu nie zależy od obecności innych cząsteczek w objętości adsorpcyjnej, dzięki czemu możliwa jest adsorpcja wielocząsteczkowa.
5. Siły adsorpcji nie zależą od temperatury, dlatego wraz ze zmianą temperatury objętość adsorpcji nie zmienia się.
Równanie Freundlicha. Powierzchnia adsorbentu jest niejednorodna, pomiędzy zaadsorbowanymi cząsteczkami zachodzi interakcja, centra aktywne nie są od siebie całkowicie niezależne. G. Freindlicha zasugerowali, że liczba moli zaadsorbowanego gazu lub substancji rozpuszczonej na jednostkę masy adsorbentu (tzw. adsorpcja właściwa X/M), powinno być proporcjonalne do ciśnienia równowagowego (dla gazu) lub stężenia równowagowego (dla substancji zaadsorbowanych z roztworu) adsorbentu podniesionego do pewnej potęgi, która jest zawsze mniejsza od jedności:
X / M = AP N X / M = AC N.
wykładniki N i współczynnik proporcjonalności A ustalone eksperymentalnie.
37. Termodynamika procesu adsorpcji. Równanie adsorpcji Gibbsa
Aby zbadać zjawisko adsorpcji na granicy „roztwór – gaz”, należy ustalić zależność pomiędzy nadmiarem zaadsorbowanej substancji w warstwie na powierzchni ( G), stężenie środka powierzchniowo czynnego w roztworze ( Z) i napięcie powierzchniowe ( σ ) na granicy faz „roztwór-gaz”. Bardziej celowe jest rozważenie zjawiska z termodynamicznego punktu widzenia i powiązanie adsorpcji substancji rozpuszczonej ze zmianą energii swobodnej powierzchni lub jej napięcia powierzchniowego. To połączenie zostało nawiązane W. Gibbsa V 1876, któremu nadano nazwę „Równanie adsorpcji Gibbsa”:
G = – Z / CZ X dσ/DC.
Nadal możesz to sobie wyobrazić Równanie Gibbsa, w oparciu o termodynamikę, wykorzystując potencjał izobaryczno-izotermiczny G, potencjały chemiczne µ 1 I μ 2 , a także używać N 1 I N 2 liczba moli składników. Po przeanalizowaniu tego biorąc pod uwagę entropię S, tom V i ciśnienie P, możemy napisać następujące równanie:
dG=– SDT+VdP+σds+ μ 1 re n 1 + μ 2 dni 2 .
Przyrównujemy to do zera, a biorąc pod uwagę stałą temperaturę i ciśnienie, upraszczamy to do równania postaci:
SD σ + n 1 d μ 1 + n2d μ 1 = 0.
Biorąc pod uwagę fakt, że dla roztworów rozcieńczonych potencjał chemiczny drugiego składnika wyraża się w następujący sposób:
μ 2 = μ 2 0 +CZ ln C,
i przy założeniu, że temperatura jest stała
dμ 2 =rtdnc,
podstawiając to równanie do
![]()
otrzymujemy pożądane równanie adsorpcji Gibbsa. Na podstawie równania widać, że jeśli napięcie powierzchniowe σ wzrasta wraz z koncentracją Z, wówczas stężenie substancji rozpuszczonej na warstwie powierzchniowej jest mniejsze niż w objętości roztworu (tzw. adsorpcja ujemna), a jeżeli napięcie powierzchniowe σ maleje wraz ze wzrostem stężenia Z, wówczas stężenie w warstwie jest większe niż w objętości (adsorpcja dodatnia) i wreszcie jeśli σ nie zależy od Z, to stężenie substancji w warstwie na powierzchni i w objętości jest takie samo. Równanie Gibbsa wyprowadzono za pomocą termodynamiki. W praktyce trudno jest zweryfikować to równanie, co wynika ze złożoności wyznaczania stężenia substancji rozpuszczonej w powierzchni warstwowej. Doświadczony B. McBena stwierdzili, że za pomocą urządzenia odcięto z powierzchni roztworu bardzo cienką warstwę cieczy. Dalsze wyznaczenie wszystkich parametrów równania Gibbsa wykazało, że ustalone eksperymentalnie wartości adsorpcji pokrywają się z wartościami obliczonymi za pomocą równania Gibbsa w granicach błędu eksperymentalnego. Ze względu na jednorodność i gładkość powierzchni dowolnej cieczy, badając adsorpcję na jej powierzchni, zwykłe wyobrażenia o centrach aktywnych są całkowicie nie do zastosowania. W temperaturze krytycznej różnica między sąsiednimi fazami zanika, a napięcie powierzchniowe z reguły staje się równe zeru. Adsorpcja gazów i par ma tak duże zastosowanie praktyczne, że w literaturze, zwłaszcza technicznej, można spotkać się z tym pojęciem, które stosowane jest jedynie w odniesieniu do procesów zachodzących na powierzchni ciał stałych.
Koncepcja ta, jak również najbardziej ogólne wzorce adsorpcji, jako rozważane równanie Gibbsa, mają zastosowanie do wszystkich granic faz. Korzystając z równania Gibbsa i wszystkich wynikających z niego zapisów, po wyznaczeniu wartości Г, można skonstruować izotermę adsorpcji.
38. Specyfika adsorpcji na materiałach mikroporowatych. Teoria potencjału Polana. Potencjał adsorpcji
Teoria Glade'a uwzględnia niezlokalizowaną adsorpcję fizyczną, która jest bezpośrednio spowodowana siłami van der Waalsa pomiędzy adsorbentem a adsorbatem (można to uznać za pierwszą pozycję). Drugim stanowiskiem tej teorii jest koncepcja pola siłowego (potencjalnego) adsorbentu, które rozciąga się na znaczną odległość od powierzchni; warstwa adsorpcyjna pojawiająca się w tym polu jest wielocząsteczkowa. Jeśli weźmiemy pod uwagę adsorpcję gazów, to gęstość tej warstwy maleje wzdłuż pewnej normalnej od powierzchni. Jeśli weźmiemy pod uwagę adsorpcję pary, wówczas na powierzchni tworzy się warstwa cieczy o określonej grubości. Pole w teorii Polanyi'ego rozpatrywane jest jako szereg powierzchni ekwipotencjalnych, każda powierzchnia odpowiada pewnej wartości potencjału ε , a każda kolejna powierzchnia będzie mniejsza od poprzedniej. Każda taka powierzchnia w przestrzeni wycina warstwy o określonej objętości, oznaczone jako v ja. Zadaniem teorii Polanyi'ego jest znalezienie przejścia od zwykłych współrzędnych izotermy ( x, s) do parametrów pola ε ja I v ja, z dalszym ustaleniem powiązania między tymi głównymi parametrami. Pierwsza część problemu, którą postawił Polanyi, jest dość skomplikowana i w wielu przypadkach nie może mieć określonych rozwiązań, ale w przypadku adsorpcji pary ta część problemu jest rozwiązywana w pierwszym przybliżeniu bardzo prosto. W przypadku warstwy adsorpcyjnej cieczy wypełniona część objętości będzie równa:
v ja \u003d x (M / d),
Gdzie D jest gęstością substancji w stanie ciekłym.
W swojej teorii M. Polyany wprowadza kolejny zapis dotyczący braku tzw. przesiewanie pola w procesie adsorpcji, wartość ε w tej teorii przestrzeni jest to wartość stała (coś w rodzaju potencjału grawitacyjnego) niezależnie od tego, czy pomiędzy danym punktem a powierzchnią stałą znajdują się pewne cząsteczki adsorbatu, czy też cała przestrzeń jest wolna. Polyani wprowadza tę koncepcję potencjał adsorpcji ε , czyli izotermiczna praca sprężania pary, gdy jest ona przenoszona spod ciśnienia równowagowego R w fazie masowej daleko od powierzchni do obszaru warstwy powierzchniowej o ciśnieniu pary nasyconej p 0 wówczas wyrażenie określające potencjał będzie wyglądać następująco:
ε = CZ ln R 0 / R.
Za pomocą takiego równania można przejść od współrzędnych x, p do współrzędnych ε I w i uzyskaj krzywą, która nazywa się „charakterystyczną”. Polanyi odkrył w swoich eksperymentach, że takie krzywe, zbudowane z danych eksperymentalnych otrzymanych izoterm, mają następującą właściwość: są niezmienne względem T, czyli innymi słowy, wszystkie krzywe tego typu mogą leżeć na jednej krzywej ε −ε .
M. Polyany przyjął to stanowisko jako postulat, tj.:
Wskazana właściwość Polyany ma ogromną wartość praktyczna, może skonstruować rodzinę izoterm na podstawie jednej eksperymentalnej izotermy adsorpcji.
Teoria Polanyi'ego nie daje analitycznego wyrażenia izotermy ani funkcji potencjału w funkcji objętości, ale pozwala obliczyć współrzędne dla dowolnej temperatury, jeśli znana jest co najmniej jedna izoterma. Wynik ten jest dla nas bardzo ważny obliczenia technologiczne, ponieważ dla podobnych gazów na tym samym adsorbencie krzywe adsorpcji mogą być blisko siebie i w wielu przypadkach mogą się nakładać.
39. Charakterystyka adsorpcji. Niezmienniczość temperaturowa i powinowactwo krzywych charakterystycznych
Pole siłowe pojawiające się na powierzchni adsorbentu może być pod wieloma względami podobne do pola grawitacyjnego. W polu adsorpcji można przedstawić powierzchnie potencjalne, czyli takie, dla których charakterystyczny jest ten sam potencjał adsorpcyjny. Zgodnie z koncepcją potencjału adsorpcyjnego θ należy rozumieć jako nic innego jak pracę wykonaną przeciw siłom adsorpcji podczas przemieszczania 1 mola adsorbatu z określonego punktu pola do określonej fazy gazowej. Maksymalny potencjał adsorpcji będzie występował na granicy „adsorbent – objętość adsorpcji”. Jednak na granicy „objętość – faza gazowa” (w tym miejscu kończy się działanie sił adsorpcji) potencjał adsorpcji musi być równy zeru. Zmianę potencjału adsorpcji wraz ze zmianą objętości adsorpcji można przedstawić w postaci krzywych. Po raz pierwszy dokonał tego M. Polyani. Tego typu krzywe nie zależą od temperatury i mogą być charakterystyczne dla każdego konkretnego adsorbentu; tego typu krzywe nazywane są zwykle charakterystycznymi krzywymi adsorpcji. Teoria adsorpcji wielocząsteczkowej zakłada, że równanie stanu gazu odnosi się do wielkości adsorpcji. W konsekwencji izotermy charakteryzujące zależność gęstości adsorbatu od objętości dla różnych temperatur przypominają izotermy zależności ciśnienia od objętości. W niskich temperaturach siły adsorpcji działające na powierzchnię mogą powodować kondensację pary w ciecz o określonej gęstości. W temperaturach niższych od krytycznej podczas kondensacji cała objętość adsorpcji zostanie wypełniona cieczą. W tym przypadku krzywa adsorpcji będzie przebiegać niemal równolegle do osi odciętej, co jest związane z małą ściśliwością cieczy. Następnie krzywa adsorpcji na granicy „objętość – faza gazowa” gwałtownie spada w dół i odpowiednio gęstość adsorbatu osiąga wartość pewnej gęstości fazy gazowej. W temperaturach wyższych od krytycznych adsorbent może zachowywać się jak gaz doskonały, a wykres będzie wyrażony jako izoterma zależności dla gazu doskonałego, pod warunkiem, że pV = CZ. W takich warunkach zaadsorbowany gaz będzie miał maksymalną gęstość na samej powierzchni adsorbentu, a minimalną w bezpośrednim sąsiedztwie fazy gazowej. Ponadto w tym przypadku należy zauważyć, że gęstość adsorbatu w warstwie adsorpcyjnej nigdzie nie osiąga gęstości samej cieczy. A jeśli temperatura będzie bardzo bliska krytyczna, zależność gęstości od objętości zostanie wyrażona krzywą zbliżoną wyglądem do izotermy opisanej Równanie van der Waalsa. W tym scenariuszu część zaadsorbowanej substancji będzie w zaadsorbowanej objętości w stanie ciekłym, a część zaadsorbowanej substancji będzie w stanie gazowym. Wtedy krzywa obniży się najostrzej w odcinku odpowiadającym przejściu ze stanu ciekłego do gazowego. Jeśli z eksperymentalnej izotermy adsorpcji jednego z adsorpcji zbuduje się krzywą charakterystyczną i znając odpowiadające jej współczynniki powinowactwa dla innego adsorbentu, można znaleźć izotermę adsorpcji i skonstruować ją dla innego adsorbentu. Potencjalna teoria adsorpcji pozwala obliczyć różne izotermy adsorpcji różnych par na tym samym adsorbencie, ponadto wykorzystując krzywą charakterystyczną otrzymaną z izotermy adsorpcji jednej pary, ponieważ stosunek potencjału adsorpcji nie zależy od objętości adsorpcji .
podobieństwo(z łac. affinis - „pokrewny”) - chromatografia powinowactwa. Metoda oczyszczania i rozdziału białek opiera się na ich selektywnym oddziaływaniu z ligandem kowalencyjnie związanym z obojętnym nośnikiem (chromatografia powinowactwa). Pomiar powinowactwa substancji toksycznej do receptora jest w rzeczywistości eksperymentalnym badaniem zależności pomiędzy ilością substancji dodanej do pożywki inkubacyjnej a ilością kompleksu substancja toksyczna-receptor powstałego w wyniku interakcji.
Termodynamika procesów adsorpcji.
| Nazwa parametru | Oznaczający |
| Temat artykułu: | Termodynamika procesów adsorpcji. |
| Rubryka (kategoria tematyczna) | Edukacja |
Podstawowe definicje i metody klasyfikacji procesów adsorpcji.
Adsorpcja odnosi się do zjawisk zachodzących w wyniku samoistnego spadku energii powierzchniowej.
Adsorpcja- proces spontanicznej odwracalnej lub nieodwracalnej redystrybucji składników układu heterogenicznego pomiędzy warstwą powierzchniową a objętością fazy jednorodnej.
W układach wieloskładnikowych zamiast warstwy wierzchniej preferowany jest składnik obniżający napięcie międzyfazowe. W układach jednoskładnikowych podczas tworzenia warstwy powierzchniowej zmienia się jej struktura (pewna orientacja atomów i cząsteczek, polaryzacja), tzw. autoadsorpcja.
Faza gęstsza, na której zlokalizowane są oddziaływania adsorpcyjne, nazywana jest fazą gęstszą adsorbent. Substancję rozdzieloną pomiędzy objętość fazy jednorodnej a warstwę powierzchniową oznaczono terminem „” adsorbatʼʼ.
W niektórych przypadkach proces adsorpcji jest odwracalny. W takim przypadku w określonych warunkach część zaadsorbowanych cząsteczek może przejść z warstwy powierzchniowej do objętości fazy w wyniku molekularnych zjawisk kinetycznych. Odwrotny proces adsorpcji nazywa się desorpcja.
Metody klasyfikacji procesów adsorpcji.
Klasyfikacja procesów adsorpcji ze względu na stan skupienia oddziałujących faz. Biorąc pod uwagę zależność od stanu skupienia faz sąsiednich, wyróżnia się następujące typy procesów adsorpcji:
Adsorpcja gazów na stałych adsorbentach;
Adsorpcja substancji rozpuszczonych na granicy faz „ciało stałe-ciecz” i „ciecz-ciecz”;
Adsorpcja środków powierzchniowo czynnych na granicy faz „ciecz-gaz”.
Klasyfikacja procesów adsorpcji ze względu na mechanizm oddziaływania adsorbentu i adsorbatu. Adsorpcję można rozpatrywać jako oddziaływanie cząsteczek adsorbatu z centrami aktywnymi adsorbentu. Ze względu na mechanizm ich oddziaływania wyróżnia się następujące rodzaje adsorpcji:
1) adsorpcja fizyczna (molekularna).- oddziaływanie cząsteczek adsorbatu z adsorbentem odbywa się pod wpływem sił van der Waalsa, wiązań wodorowych (bez reakcje chemiczne);
2) adsorpcja chemiczna (chemisorpcja)– przyłączenie cząsteczek adsorbatu do miejsc aktywnych adsorbentu następuje w wyniku różnego rodzaju reakcji chemicznych (z wyjątkiem reakcji wymiany jonowej);
3) Adsorpcja jonowymienna (wymiana jonowa) - redystrybucja substancji adsorbatu pomiędzy roztworem a fazą stałą (wymiennik jonowy) zgodnie z mechanizmem reakcji wymiany jonowej.
Do ilościowego opisu procesów adsorpcji stosuje się dwie wielkości.
1) Absolutna adsorpcja oznacza ilość (mol) lub masę (kg) adsorbatu na jednostkę powierzchni lub masę adsorbentu. Oznaczenie - A; wymiar: mol / m 2, mol / kg, kg / m 2, kg / kᴦ.
2) Adsorpcja Gibbsa (nadmiarowa). to nadmiar substancji adsorbatu w warstwie powierzchniowej o określonej grubości w stosunku do jej ilości w objętości fazy jednorodnej, na jednostkę powierzchni lub masy adsorbentu. Oznaczenie - G; jednostka: mol/m 2 , mol/kᴦ.
Zależność pomiędzy adsorpcją bezwzględną i nadmiarową można zilustrować równaniem:
G \u003d A - c * h (3,1)
gdzie c jest równowagowym stężeniem substancji w objętości fazy, mol/m3;
h jest grubością warstwy wierzchniej, warunkowo przyjmowaną równą 10-9 m.
W wieloskładnikowych układach heterogenicznych, gdy jeden lub drugi składnik jest redystrybuowany pomiędzy objętością fazy jednorodnej a warstwą powierzchniową, obowiązuje równanie na nadmiar energii wewnętrznej powierzchni:
U = T * S + s * s + Sm i * n ja (3.2)
Doprowadzając wszystkie warunki równania do pola jednostkowego powierzchni międzyfazowej, otrzymujemy:
U s = T * S s + s + Sm i * Г ja (3.3)
gdzie Г i = n i / s jest nadmiarem i-tego składnika w warstwie powierzchniowej, czyli adsorpcją Gibbsa.
Dla układu jednoskładnikowego równanie (3.3) przyjmuje postać:
G s = s + m * Г (3.4)
gdzie G s = U s - T * S s to energia Gibbsa powierzchni lub praca tworzenia jednostki powierzchni;
m * G - zagęszczenie substancji zaadsorbowanej w warstwie powierzchniowej.
Na podstawie równania (3.4) można stwierdzić, że podczas adsorpcji praca nad utworzeniem powierzchni międzyfazowej polega na pracy polegającej na uformowaniu powierzchni (rozerwaniu wiązań kohezyjnych w większości fazy adsorbatu) i zagęszczeniu substancji w warstwie powierzchniowej.
W stanie równowagi dynamicznej pomiędzy adsorbentem i adsorbatem, przy zmianie energii Gibbsa układu heterogenicznego ΔG = 0, termodynamikę procesu adsorpcji opisuje równanie zwane Podstawowe równanie adsorpcji Gibbsa:
Ds = S° i * dm i (3,5)
Równanie to jest uniwersalne, ponieważ obowiązuje dla wszystkich typów procesów adsorpcji
Szczególne przypadki równania adsorpcji Gibbsa.
1) Adsorpcja z roztworów.
Dla potencjału chemicznego i-tego składnika układu podczas adsorpcji na granicy faz „ciecz – ciało stałe” i „ciecz – gaz” obowiązują równania:
m ja = m ja 0 + R*T*ln a ja (3.6)
dm i = R*T* d ln a i (3.7)
gdzie m i 0 jest potencjałem chemicznym i-tego składnika układu w standardowe warunki;
a i – aktywność i-tego komponentu systemu w warunkach standardowych.
Na tej podstawie równanie adsorpcji Gibbsa przyjmie postać:
Г i = - a i / R*T * (ds / da i) (3.8)
W przypadku roztworów nieelektrolitowych bierzemy a i \u003d c i, następnie:
Г i \u003d - s / R * T * (ds / ds) (3,9)
Dla roztworów elektrolitów:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
gdzie c ± jest średnim stężeniem jonowym roztworu;
n jest współczynnikiem stechiometrycznym.
2) Adsorpcja substancji z fazy gazowej.
Zgodnie z równaniem Mendelejewa-Claiperona:
P \u003d c * R * T (3,11)
W związku z tym równanie Gibbsa dotyczące adsorpcji gazów na stałych adsorbentach zapisuje się w następującej formie:
Г i = - Р / R*T * (ds / dР) (3.12)
W praktyce równanie adsorpcji Gibbsa umożliwia obliczenie wielkości adsorpcji substancji w warstwie międzyfazowej, dla której wyznacza się napięcie powierzchniowe, na podstawie pomiaru napięcia powierzchniowego przy różnych wartościach stężenia cieczy lub równowagowego ciśnienia gazu .
Termodynamika procesów adsorpcji. - koncepcja i rodzaje. Klasyfikacja i cechy kategorii „Termodynamika procesów adsorpcji”. 2017, 2018.
W przypadku oddziaływania dwóch atomów:
U jest energią interakcji;
U = U + CIĄGNIJ.
 - Równanie Lennarda-Jonesa
, do, b, m = stała
- Równanie Lennarda-Jonesa
, do, b, m = stała
W przypadku oddziaływania atomów z powierzchnią stałą konieczne jest zsumowanie wszystkich oddziaływań.
x to odległość do powierzchni
r - promień działania sił przyciągania
dV - objętość
n to liczba cząsteczek powierzchniowych
U REKLAMY. jest energią interakcji adsorpcji



W przypadku adsorpcji następuje zwiększenie przyciągania. Natomiast w przypadku interakcji typu niepolarno-niepolarnego, adsorpcja jest zlokalizowana głównie w zagłębieniach.
oddziaływanie elektrostatyczne.
Adsorbent polarny - adsorbat niepolarny
Adsorbent niepolarny - adsorbat polarny
Adsorbent polarny to adsorbat polarny.
M  cząsteczka adsorbatu jest reprezentowana jako dipol, a adsorbent jako przewodnik, w którym cząsteczka adsorbatu indukuje dipol zwierciadlany symetrycznie względem danego.
cząsteczka adsorbatu jest reprezentowana jako dipol, a adsorbent jako przewodnik, w którym cząsteczka adsorbatu indukuje dipol zwierciadlany symetrycznie względem danego.
X - odległość do środka
Podczas interakcji pojawia się potencjał:
 ,
,
 jest moment dipolowy.
jest moment dipolowy.
Potencjał ma tendencję do przyjmowania wartości maksymalnej, tj. dipole mają tendencję do ustawiania się prostopadle do powierzchni.
Ponieważ wzrost temperatury sprzyja wzrostowi ruchów Browna, prowadzi to do spowolnienia procesu adsorpcji.
W przypadku oddziaływań elektrostatycznych adsorbat lokalizuje się głównie na występach.
Podstawowe równanie adsorpcji.
W przypadku adsorpcji składnik ulega redystrybucji, co oznacza, że zmienia się potencjał chemiczny. Proces adsorpcji można uznać za przemianę energii powierzchniowej w energię chemiczną.
Objętość warstwy = 0, następnie uogólnione równanie I i II zasady termodynamiki:
T = stała; (1) = (2) => 

Dla układu dwuskładnikowego:
,
 ,
,
 =>
=>
=>
 - Równanie adsorpcji Gibbsa
.
- Równanie adsorpcji Gibbsa
.
W przypadku adsorpcji TV. korpus - gaz:, 
 ,
,

 - izoterma
- izoterma
 - izobar
- izobar
 - izopikne
- izopikne
 - izoster
- izoster
Izoterma, izopikne, izostera są ze sobą powiązane.
Ponieważ funkcja adsorpcji 



Izoterma Henry'ego Izoterma Langmuira



Termodynamika. Adsorpcja.
W przypadku mediów skondensowanych:
 ,
,
 ,
,
 - całkowa zmiana energii Gibbsa
.
- całkowa zmiana energii Gibbsa
.

Ciśnienie P na zakrzywionej powierzchni, ciśnienie P S na płaskiej powierzchni
 - potencjał adsorpcji
- potencjał adsorpcji
Różnicowa zmiana entrapii 
 , Г = stała
, Г = stała
- różnicowa zmiana entropii
- różnicowa entalpia adsorpcji
 - izosteryczne ciepło adsorpcji
- izosteryczne ciepło adsorpcji
 - ciepło kondensacji
- ciepło kondensacji
 - ciepło netto adsorpcji
- ciepło netto adsorpcji

 ,
,




Qa jest całkowitym ciepłem adsorpcji, 
Qra jest całkowitym ciepłem netto adsorpcji,

Równanie Henry'ego
Badanie adsorpcji utrudnia niejednorodność powierzchni, dlatego najprostsze prawidłowości uzyskuje się dla powierzchni jednorodnych.
Rozważmy oddziaływanie gazów z powierzchnią stałą, gdy gaz przechodzi ze stanu równowagi objętościowej do stanu równowagi na powierzchni. Przypadek ten jest analogiczny do równowagi gazów w polu grawitacyjnym.
 ,
,
 ,
=>
,
=> -Równanie Henry'ego
-Równanie Henry'ego
 - współczynnik podziału
- współczynnik podziału
W procesie adsorpcji następuje zmiana potencjałów chemicznych.
Dla fazy masowej: 
Dla gazu powierzchniowego: 
W stanie równowagi  , tj.
, tj.

W równaniu Henry'ego stała nie zależy od stężenia
Równanie Henry'ego obowiązuje w obszarze niskich ciśnień i stężeń. Wraz ze wzrostem stężenia możliwe są 2 rodzaje odchyleń od prawa Henry'ego:
1 - odchylenia dodatnie, D maleje, A maleje
2 - odchylenia ujemne, D - wzrosty, A - wzrosty.
Rodzaj odchylenia zależy od przewagi jednego lub drugiego rodzaju interakcji adsorbent-adsorbat.
Przy silnym oddziaływaniu kleju współczynniki aktywności wzrastają - odchylenie dodatnie. W przypadku oddziaływań kohezyjnych obserwuje się odchylenia ujemne.
adsorpcja monocząsteczkowa.
Izoterma Langmuira.
Najprostsze prawidłowości uzyskano w teorii Henry’ego. Langmuir zaproponował teorię, zgodnie z którą adsorpcję uważa się za reakcję quasi-chemiczną. W której:
Powierzchnia jest energetycznie jednolita.
Adsorpcja jest zlokalizowana, każde centrum adsorpcji oddziałuje z jedną cząsteczką adsorbatu.
Cząsteczki adsorbatu nie oddziałują ze sobą.
Adsorpcja jest jednowarstwowa.

 - powierzchnia,
- powierzchnia,  - adsorbat,
- adsorbat,  - kompleks adsorpcyjny.
- kompleks adsorpcyjny.
 , to stężenie miejsc adsorpcji:
, to stężenie miejsc adsorpcji:  ,
, - ograniczenie adsorpcji.
- ograniczenie adsorpcji.
 , wówczas stała reakcji:
, wówczas stała reakcji: 

 - Równanie Langmuira.
- Równanie Langmuira.
Adsorpcja a stężenie
1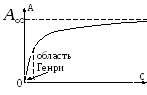 )
)
 ,
,
2) obszar wysokich stężeń
 - ograniczenie adsorpcji, tworzenie warstwy monocząsteczkowej
- ograniczenie adsorpcji, tworzenie warstwy monocząsteczkowej
Dla energii Gibbsa: .
g jest współczynnikiem entropii.
W przypadku izotermy Henry'ego energia Gibbsa charakteryzuje przejście adsorbatu ze stanu standardowego w masie do stanu standardowego na powierzchni. W przypadku izotermy Langmuira  charakteryzuje stopień powinowactwa adsorbentu i adsorbatu.
charakteryzuje stopień powinowactwa adsorbentu i adsorbatu.
 znalezione z izobary van't Hoffa.
znalezione z izobary van't Hoffa.

 , Następnie
, Następnie  , stąd
, stąd  .
.

 - stopień wypełnienia powierzchni.
- stopień wypełnienia powierzchni.




 - ilość wolnych miejsc pracy,
- ilość wolnych miejsc pracy,  - liczba zajętych miejsc.
- liczba zajętych miejsc.
 ,
,

Te. w obszarze wysokich stężeń liczba wolnych miejsc jest odwrotnie proporcjonalna do ilości adsorbatu.
Adsorpcja mieszaniny gazów na jednorodnej powierzchni.
W tym przypadku proces adsorpcji uważa się za dwie równoległe reakcje.
 (1)
(1)

 (2)
(2)


Adsorpcja mieszaniny gazów na niejednorodnej powierzchni.
W przypadku niejednorodnej powierzchni nie należy ograniczać się do wypełnień średnich.
W wyniku konkurencji możliwa jest lokalizacja różnych adsorbatów w różnych typach lokalizacji.
W tym wypadku relacja  .
.
 ,
,
 jest prężnością pary nasyconej adsorbatu.
jest prężnością pary nasyconej adsorbatu.
 ,
,
 jest ciepłem adsorpcji.
jest ciepłem adsorpcji.
„+” – zależność symbatyczna, „-” – zależność antybakteryjna, „H” – brak korelacji.
„+” - adsorpcja przebiega według tego samego mechanizmu. W najbardziej korzystnych energetycznie obszarach adsorbowany jest głównie gaz o dużym powinowactwie do powierzchni.
„-” - adsorpcja przebiega poprzez różne mechanizmy i do pewnego momentu nie ma już konkurencji o powierzchnię.
Adsorpcja monocząsteczkowa realizowana jest głównie podczas fizycznej adsorpcji gazów przy niskich wartościach P, jak również na granicy faz ciecz/gaz.
Adsorpcja wielocząsteczkowa.
Teoria zakładu(Brunauer, Emmet, Teller).
W przypadku, gdy utworzenie monowarstwy nie jest wystarczające do skompensowania energii powierzchniowej, adsorpcja ma charakter wielocząsteczkowy i można ją rozpatrywać w wyniku wymuszonej kondensacji pod działaniem sił powierzchniowych.
Podstawowe postanowienia:
Kiedy cząsteczka adsorbatu uderza w zajęte miejsce, powstaje zestaw wielokrotny.
Gdy się zbliżysz P Do P S zmniejsza się liczba miejsc wolnej adsorpcji. Początkowo liczba miejsc zajmowanych przez osoby samotne, podwójne itp. wzrasta, a następnie maleje. zestawy.
Na P =P S adsorpcja zamienia się w kondensację.
Nie ma interakcji poziomych.
Dla pierwszej warstwy wykonywana jest izoterma Langmuira.
Powierzchnię traktuje się jako zbiór miejsc adsorpcji. Obowiązuje warunek równowagi dynamicznej: szybkość kondensacji w wolnych miejscach jest równa szybkości parowania z zajętych.
a jest współczynnikiem kondensacji (ułamek cząsteczek skondensowanych na powierzchni);
 ,
,
Zm to maksymalna liczba wolnych miejsc.
 - częstotliwość drgań atomów w kierunku prostopadłym do powierzchni.
- częstotliwość drgań atomów w kierunku prostopadłym do powierzchni.
Dla pierwszej warstwy warunki równowagi dynamicznej są następujące:



 , Następnie
, Następnie
 - Równanie Langmuira.
- Równanie Langmuira.
Dla drugiej warstwy będzie prawdą: 
Dla i-tej warstwy: 
Dla uproszczenia przyjmuje się, że a i ν są takie same dla wszystkich warstw z wyjątkiem pierwszej. Dla wszystkich warstw z wyjątkiem pierwszej ciepło adsorpcji jest stałe. Dla ostatniej warstwy ciepło adsorpcji jest równe ciepłu kondensacji. W rezultacie równanie
 (*)
(*)
C- stała,
W przypadku teorii BET stała Z charakteryzuje energię Gibbsa czystej adsorpcji. Równanie zawiera tylko jedną stałą i równanie to jest również bardzo ważne przy określaniu powierzchni właściwej adsorbentu.
Ponieważ w wyniku adsorpcji wydziela się ciepło, wyznaczanie powierzchni właściwych przeprowadza się w niskich temperaturach.
 ????????????
????????????


Główny błąd teorii– Zaniedbanie oddziaływań poziomych na rzecz wertykalnych.
Równanie mieści się w przedziale  od 0,05 do 0,3.
od 0,05 do 0,3.
Gdzie  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 - wpływa oddziaływanie adsorbat - adsorbat.
> 0,3 - wpływa oddziaływanie adsorbat - adsorbat.
Uwzględnianie oddziaływań adsorbat-adsorbat.
Interakcje pojawiają się podczas adsorpcji na niepolarnej powierzchni rozgałęzionych cząsteczek lub cząsteczek. zdolne do tworzenia stowarzyszeń. W tym przypadku zmienia się kształt izoterm adsorpcji.
A  sorbent nie jest polarny.
sorbent nie jest polarny.
Wykres 1 odpowiada oddziaływaniom słabym adsorbat-adsorbat, mocny adsorbat-adsorbent.
Wykres 2 odpowiada silnemu oddziaływaniu adsorbat-adsorbat, silnemu oddziaływaniu adsorbat-adsorbent.
Wykres 3 przedstawia silne oddziaływanie adsorbat-adsorbat, słabe oddziaływanie adsorbat-adsorbent.
 ,
,

W przypadku interakcji pomiędzy cząsteczkami adsorbatu należy uwzględnić zmiany współczynników aktywności. A to równanie zapisuje się jako:
 - równanie Frunkina, Fowlera, Guggenheima.
- równanie Frunkina, Fowlera, Guggenheima.
k jest stałą przyciągania.
Teoria potencjału Polana.
Teoria ta nie wyprowadza żadnego rodzaju izotermy adsorpcji, ale umożliwia obliczenie izoterm w innej temperaturze.
Adsorpcja jest wynikiem przyciągania adsorbatu do powierzchni adsorbentu pod wpływem działania potencjału adsorpcyjnego, który nie zależy od obecności innych cząsteczek, a zależy od odległości powierzchni od cząsteczki adsorbatu.
 ,
,
 - potencjał adsorpcji.
- potencjał adsorpcji.
Ponieważ powierzchnia jest niejednorodna, odległość zastępuje się objętością adsorpcji  .objętość adsorpcji jest objętością zawartą pomiędzy powierzchnią a punktem odpowiadającym danej wartości
.objętość adsorpcji jest objętością zawartą pomiędzy powierzchnią a punktem odpowiadającym danej wartości  .
.
Potencjał adsorpcji to praca przeniesienia 1 mola adsorbatu poza zadaną objętość adsorpcji do zadanego punktu objętości adsorpcji (lub praca przeniesienia 1 mola pary nasyconej adsorbatu, który jest w równowadze z ciekłym adsorbatem przy braku adsorbentu do fazy gazowej znajdującej się w równowadze z adsorbentem).

Krzywa charakterystyczna
 - potencjał adsorpcji,
- potencjał adsorpcji, 
Dla danego adsorbentu i różnych adsorbatów prawdziwe jest stwierdzenie: 
Dla różnych typów adsorbatów  ,
,
Gdzie  potencjały izoterm adsorpcji przy ciśnieniach względnych
potencjały izoterm adsorpcji przy ciśnieniach względnych  dla adsorbatu 1 i dla adsorbatu 2. Stosunek ten jest wartością stałą.
dla adsorbatu 1 i dla adsorbatu 2. Stosunek ten jest wartością stałą.
 - współczynnik powinowactwa
- współczynnik powinowactwa
Teoria kondensacji kapilarnej.
Przebieg procesu adsorpcji w dużej mierze zależy od budowy ciała porowatego.
|
mikroporowaty | |
|
Przejściowy porowaty | |
|
Makroporowaty |
W przypadku sorbentów mikroporowatych pola sił adsorpcji nakładają się. W przypadku sorbentów makroporowatych pory pełnią rolę kanałów transportowych. Procesy kondensacji są najbardziej znaczące w przejściowych ciałach porowatych. Kondensacja kapilarna rozpoczyna się przy określonych wartościach P I  gdy część energii powierzchniowej została już skompensowana. Warunkiem koniecznym jest to, aby powierzchnia była samonośna. Proces jest opisany Równanie Thompsona-Kelvina.
gdy część energii powierzchniowej została już skompensowana. Warunkiem koniecznym jest to, aby powierzchnia była samonośna. Proces jest opisany Równanie Thompsona-Kelvina.

 - w przypadku zwilżania środek krzywizny znajduje się w fazie gazowej.
- w przypadku zwilżania środek krzywizny znajduje się w fazie gazowej.
W przypadku kondensacji kapilarnej izoterma adsorpcji ma postać histerezy. Dolna gałąź odpowiada procesowi adsorpcji, a górna gałąź odpowiada procesowi desorpcji.
Wszystkie rodzaje porów można zredukować do trzech typów:
|
stożkowy |
Cylindryczny z jednym zamkniętym końcem |
Cylindryczny z dwoma otwartymi końcami |
|
Wypełnianie procesowe odbywa się od dołu porów. Isterma adsorpcji i izoterma desorpcji w tym przypadku pokrywają się, ponieważ proces adsorpcji rozpoczyna się od kuli, a proces desorpcji rozpoczyna się również wraz ze zniknięciem niektórych kul.
↓ |
Nie ma histerezy. Skok do przodu i do tyłu opisuje równanie:
|
Nigdzie nie ma dna, wypełnienie porów będzie przebiegać wzdłuż ścian cylindra.
cylinder: Izoterma i będzie miała postać histerezy.
↓ |


 - kula,
- kula, ,
,



W  warunkach zwilżania, przy niższych ciśnieniach następuje kondensacja, co jest korzystne energetycznie. Z gałęzi desorpcji otrzymuje się krzywe rozkładu wielkości porów.
warunkach zwilżania, przy niższych ciśnieniach następuje kondensacja, co jest korzystne energetycznie. Z gałęzi desorpcji otrzymuje się krzywe rozkładu wielkości porów.
Maksimum krzywej różniczkowej jest przesunięte w lewo względem punktu przegięcia całki. Całkowita objętość małych porów jest niewielka, ale mają duże powierzchnie. Wraz ze wzrostem wielkości porów zwiększa się ich objętość  , a obszar jako
, a obszar jako  , dzięki temu obserwuje się przesunięcie maksimum krzywej różniczkowej.
, dzięki temu obserwuje się przesunięcie maksimum krzywej różniczkowej.
Adsorpcja na granicy faz ciało stałe-ciecz.
W przypadku adsorpcji na granicy faz ciało stałe-gaz zaniedbaliśmy jeden składnik. W przypadku adsorpcji na granicy faz ciało stałe-ciecz adsorbat wypiera cząsteczki rozpuszczalnika z powierzchni adsorbentu.
 ,
,
Prawidłowe równanie to:

 ,
,
N 1, N 2 - ułamki molowe rozpuszczalnika i składnika, N 1 + N 2 \u003d 1, następnie
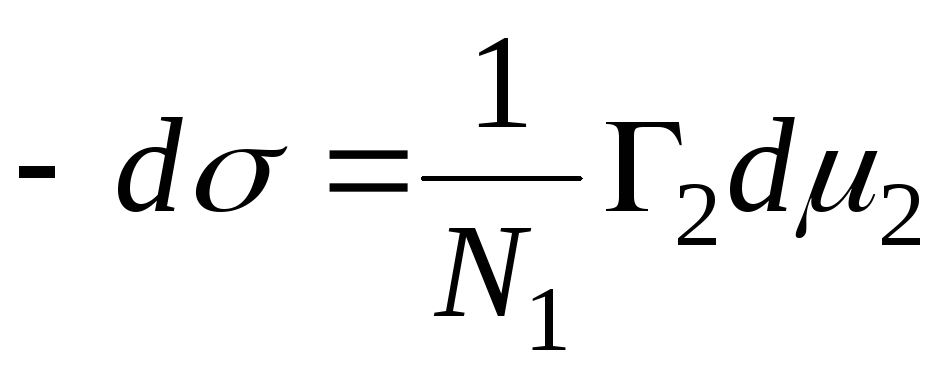 ,
=>
,
=>
 , następnie - równanie adsorpcji dla granicy faz ciało stałe - ciecz.
, następnie - równanie adsorpcji dla granicy faz ciało stałe - ciecz.
Adsorpcja (G) > 0 at  <
0
<
0
Jeśli wartości  dla składnika i rozpuszczalnika są bardzo różne, w tym przypadku zależność G z N ma ekstremum przy tej wartości N
~ 0,5.
dla składnika i rozpuszczalnika są bardzo różne, w tym przypadku zależność G z N ma ekstremum przy tej wartości N
~ 0,5.
mi  Jeśli
Jeśli  mają podobne wartości, w tym przypadku znak adsorpcji może się zmienić. Uzależnienie G z N przecina oś x
mają podobne wartości, w tym przypadku znak adsorpcji może się zmienić. Uzależnienie G z N przecina oś x
Przecięcie funkcji G(N) z osią odciętych azeotrop adsorpcyjny. Oznacza to, że na tym adsorbencie nie można rozdzielić obu składników.
Równanie izotermy adsorpcji ze stałą wymiany.
Podczas adsorpcji na granicy faz ciało stałe-ciecz składniki ulegają ciągłej redystrybucji pomiędzy powierzchnią adsorbentu a objętością roztworu.
 - komponenty (- - odnoszą się do powierzchni)
- komponenty (- - odnoszą się do powierzchni)

 ,
,
 ,
, .
.
 ,
,


Adsorpcja na granicy faz ciecz-gaz
R  Rozważmy zmianę profilu stężenia w wyniku przecięcia granicy faz ciecz-gaz. Niech składnik 2 będzie lotny.
Rozważmy zmianę profilu stężenia w wyniku przecięcia granicy faz ciecz-gaz. Niech składnik 2 będzie lotny.
Cs to stężenie w warstwie powierzchniowej.
Na podstawie definicji nadmiernej adsorpcji

Jeżeli składnik nie jest lotny, wartość adsorpcji zostanie zapisana w następujący sposób:
P  ri
ri 

W równaniu  naturę materii opisuje pochodna
naturę materii opisuje pochodna  .
.
Izoterma napięcia powierzchniowego może mieć postać 1 lub 2:
1 - środki powierzchniowo czynne
2 - środki powierzchniowo czynne
Aktywność powierzchniowa g to zdolność substancji do zmniejszania napięcia powierzchniowego w układzie.

 - grubość warstwy wierzchniej
- grubość warstwy wierzchniej
C S oznacza stężenie składnika w warstwie powierzchniowej
Z– stężenie objętościowe
Dla szeregu homologicznego obowiązuje zasada:
 - Rządy Traubeau Duclos
- Rządy Traubeau Duclos
Dla szeregu homologicznego izoterma adsorpcji wygląda następująco:
Piszemy D zamiast A, ponieważ adsorpcja w warstwie powierzchniowej jest nadmierna.
Izoterma napięcia powierzchniowego:

 jest napięciem powierzchniowym czystego rozpuszczalnika.
jest napięciem powierzchniowym czystego rozpuszczalnika.
 - podstawowe równanie adsorpcji;
- podstawowe równanie adsorpcji;
 - Równanie Langmuira.
- Równanie Langmuira.
Rozwiążmy je razem:

- Równanie Szyszkowskiego.
B jest stałą dla szeregu homologicznego.
A- przy przechodzeniu od jednego homologu do drugiego wzrasta 3-3,5 razy 
![]()
1 - obszar niskich stężeń
![]()
2 - średnie stężenie
3 - warstwa jednocząsteczkowa
Surfaktanty są cząsteczkami amfifilowymi, tj. obejmują grupę polarną i niepolarny rodnik węglowodorowy.
o jest polarną częścią cząsteczki.
| jest niepolarną częścią cząsteczki.
W rozpuszczalniku polarnym cząsteczki środka powierzchniowo czynnego są zorientowane w taki sposób, że polarna część cząsteczki jest zwrócona w stronę rozpuszczalnika, natomiast część niepolarna jest wypychana do fazy gazowej.
W równaniu Szyszkowskiego  , jest stała dla szeregu homologicznego.
, jest stała dla szeregu homologicznego.
Zaczyna pojawiać się działanie powierzchniowo czynne N>5. Przy stężeniach wyższych niż stężenie warstwy monocząsteczkowej w roztworach środków powierzchniowo czynnych następuje micelizacja.
Micele- nazywa się agregat amfifilowych cząsteczek środka powierzchniowo czynnego, którego rodniki węglowodorowe tworzą rdzeń, a grupy polarne przekształcają się w fazę wodną.
Masa micelarna – masa micelarna.
H  liczba cząsteczek jest liczbą agregacji.
liczba cząsteczek jest liczbą agregacji.
Micele sferyczne
W przypadku micelizacji w roztworze ustala się równowaga
CMC to krytyczne stężenie miceli.
Ponieważ uważamy micelę za oddzielną fazę:
Dla szeregu homologicznego istnieje równanie empiryczne:
A jest energią rozpuszczania grupy funkcyjnej.
B jest przyrostem potencjału adsorpcji, pracą adsorpcji na jednostkę metylenu.
jest przyrostem potencjału adsorpcji, pracą adsorpcji na jednostkę metylenu.
Obecność rdzenia węglowodorowego w micelach umożliwia rozpuszczenie związków nierozpuszczalnych w wodzie w wodnych roztworach środków powierzchniowo czynnych, zjawisko to nazywa się solubilizacją (rozpuszcza się solubilizat, środek powierzchniowo czynny jest solubilizatorem).
Błoto może być całkowicie niepolarne, może zawierać zarówno części polarne, jak i niepolarne i będzie zorientowane jak cząsteczka środka powierzchniowo czynnego.
W każdym przypadku podczas solubilizacji następuje wzrost masy micelarnej i liczby agregacji nie tylko na skutek włączenia solubilizatu, ale także na skutek wzrostu liczby cząsteczek surfaktantu niezbędnych do utrzymania stanu równowagi.
Solubilizacja jest tym skuteczniejsza, im niższa jest masa cząsteczkowa solubilizatu.

 ~ 72 mN/m.
~ 72 mN/m.
 ~ 33 mN/m.
~ 33 mN/m.
Skuteczność środków powierzchniowo czynnych zależy od wielkości CMC.
Nacisk warstwy powierzchniowej 2D

→ -siły napięcia powierzchniowego.
- dwuwymiarowe ciśnienie.
Warstwa powierzchniowa to siła równa różnicy napięć powierzchniowych roztworu środka powierzchniowo czynnego i czystego rozpuszczalnika, skierowana w stronę czystej powierzchni.
Pomiędzy roztworem a warstwą powierzchniową ustala się równowaga



Na  istnieje obszar, w którym
istnieje obszar, w którym  liniowo zależny od stężenia.
liniowo zależny od stężenia.
G [mol / m2].
 obszar zajmowany przez jeden mol substancji
obszar zajmowany przez jeden mol substancji
Wtedy dwuwymiarowa izoterma ciśnienia będzie miała postać 
 jest dwuwymiarową izotermą ciśnienia.
jest dwuwymiarową izotermą ciśnienia.
Uzależnienie  od SM:
od SM:

Na  - ciśnienie dwuwymiarowe gwałtownie wzrasta. Na
- ciśnienie dwuwymiarowe gwałtownie wzrasta. Na  dwuwymiarowość ulega deformacji, co powoduje gwałtowny wzrost
dwuwymiarowość ulega deformacji, co powoduje gwałtowny wzrost  .
.
Folia obustronnie ograniczona tymi samymi fazami nazywana jest dwustronną. W takich filmach obserwuje się ciągły ruch ługu macierzystego.
Folie o grubości mniejszej niż 5 nm nazywane są foliami czarnymi.

Warstwy adsorpcyjne powinny charakteryzować się dwiema cechami: lepkością i łatwością przemieszczania się, płynnością i elastycznością.
Efekt Marangoni ma charakter samoleczenia.
Trójkąt Gibbsa,  - nadciśnienie.
- nadciśnienie.
Folia jest rozciągana, a w związku z tym, że część cieczy zniknęła, środki powierzchniowo czynne przedostają się w wolną przestrzeń. Trójkąt Gibbsa.
Wpływ siły adsorpcji ciał.
Na powierzchni folii zawsze znajduje się warstwa adsorpcyjna, dla której wówczas
Równanie Langmuira:


 w dwuwymiarowe ciśnienie
w dwuwymiarowe ciśnienie
 - analog równania Szyszkowskiego
- analog równania Szyszkowskiego
zjawiska elektrokinetyczne. Podwójna warstwa elektryczna (DES).
Model Helemholtza. Teoria Gouya-Chapmana.
Lot 1808

U – ukształtowana rurka, zanurzone w niej 2 elektrody. Naruszone zostaje prawo naczyń połączonych i następuje zmiana poziomu cieczy w rurze – zjawiska elektrokinetyczne.
Zjawiska kinetyczne:
elektroforeza
Elektroosmoza
Potencjał przepływu (przepływu).
Potencjał sedymentacyjny
1 i 2 powstają w wyniku przyłożenia różnicy potencjałów, 3 i 4 przebijanie i sedymentacja cząstek koloidalnych powoduje pojawienie się różnicy potencjałów.
Elektroosmoza jest ruchem ośrodka dyspersyjnego względem stacjonarnej fazy rozproszonej pod wpływem prądu elektrycznego.
elektroforeza to ruch cząstek fazy rozproszonej względem stacjonarnego ośrodka dyspersyjnego pod wpływem prądu elektrycznego.
P  Przyczyną występowania zjawisk elektrokinetycznych jest przestrzenne rozdzielenie ładunków i pojawienie się podwójnej warstwy elektrycznej.
Przyczyną występowania zjawisk elektrokinetycznych jest przestrzenne rozdzielenie ładunków i pojawienie się podwójnej warstwy elektrycznej.
Elektryczna podwójna warstwa to płaski kondensator, jedna płytka jest utworzona przez jony określające potencjał, druga przez przeciwne. Jony są również zanieczyszczone, gdy kojony determinujące potencjał są wypychane do większości roztworu. Odległość między płytami  . Potencjał spada liniowo, różnica potencjałów
. Potencjał spada liniowo, różnica potencjałów  .
.
Zewnętrzna różnica potencjałów powoduje pojawienie się modułu sprężystości przy ścinaniu  jest parą sił na jednostkę powierzchni działających wzdłuż powierzchni ciała stałego.
jest parą sił na jednostkę powierzchni działających wzdłuż powierzchni ciała stałego.

W równowadze moduł ścinania jest równy modułowi tarcia lepkiego (  ).
).

W naszych warunkach  ,
,

 - Równanie Helemholtza-Smalukovsky'ego
- Równanie Helemholtza-Smalukovsky'ego
 - liniowe przesunięcie prędkości w i fazach.
- liniowe przesunięcie prędkości w i fazach.
mi jest natężeniem pola elektrycznego.
 - różnica potencjałów pomiędzy płytkami
- różnica potencjałów pomiędzy płytkami
 - ruchliwość elektroforetyczna [m 2 /(V * s)].
- ruchliwość elektroforetyczna [m 2 /(V * s)].
Model Helemholtza nie uwzględnia ruchu termicznego cząsteczek. W rzeczywistości rozkład jonów w warstwie podwójnej jest bardziej złożony.
Gouy i Chapman zidentyfikowali następujące przyczyny DES:
Przejście jonu z jednej fazy do drugiej po ustaleniu równowagi.
Jonizacja materii fazy stałej.
Uzupełnianie powierzchni jonami obecnymi w ośrodku dyspersyjnym.
Polaryzacja z zewnętrznego źródła prądu.
Podwójna warstwa elektryczna ma strukturę rozmytą lub rozproszoną. Jony są zwykle równomiernie rozmieszczone w warstwie rozproszonej.
Warstwa rozproszona składa się z przeciwjonów, długość warstwy zależy od ich energii kinetycznej. W temperaturze zmierzającej do zera absolutnego przeciwne są jak najbliżej jonów determinujących potencjał.
Teoria ta opiera się na dwóch równaniach:
Równanie Boltzmanna

 - przeciwdziałać siłom oddziaływania elektrostatycznego.
- przeciwdziałać siłom oddziaływania elektrostatycznego.
 jest gęstością ładunku nasypowego.
jest gęstością ładunku nasypowego.

Równanie Poissona 
Ponieważ grubość DEL jest znacznie mniejsza niż wielkość cząstek, a dla płaskiego DEL pochodna po współrzędnych  I
I  zostaje zniesiony.
zostaje zniesiony. 
Dla e y z y<<1 функцию можно разложить в ряд Маклорена:

Ograniczamy się zatem do dwóch członków szeregu:


 - Grubość DEL to odległość, o jaką maleje potencjał DEL mi raz.
- Grubość DEL to odległość, o jaką maleje potencjał DEL mi raz.
Im niższa temperatura, tym mniej  . W Т →0 – płaska DES. Im wyższe stężenie, tym więcej I, tym mniej
. W Т →0 – płaska DES. Im wyższe stężenie, tym więcej I, tym mniej  .
.





 „–” oznacza, że potencjał maleje wraz z odległością. =>
„–” oznacza, że potencjał maleje wraz z odległością. =>
=>

 ,
,
 - potencjał maleje wykładniczo.
- potencjał maleje wykładniczo.
Potencjał gęstości ładunku powierzchniowego: 
Ładunek powierzchniowy to ładunek kosmiczny o przeciwnym znaku, całkowany ze względu na odległość.




=>


Gdzie potencjał zmniejsza się 2,7 razy - 
Pojemność podwójnej warstwy 
Wadą teorii jest to, że nie uwzględnia się obecności warstwy Helemholtza, tj. nie bierze pod uwagę  , stąd błędy w określeniu głównych parametrów. Nie wyjaśnia również wpływu jonów o różnym charakterze na grubość podwójnej warstwy elektrycznej.
, stąd błędy w określeniu głównych parametrów. Nie wyjaśnia również wpływu jonów o różnym charakterze na grubość podwójnej warstwy elektrycznej.
Teoria Sterna. Struktura miceli koloidalnej.
Podwójna warstwa elektryczna składa się z dwóch części: gęstej i rozproszonej. W wyniku oddziaływania jonów tworzących potencjał ze specyficznie zaadsorbowanymi jonami powstaje gęsta warstwa. Jony te z reguły są częściowo lub całkowicie odwodnione i mogą mieć taki sam lub przeciwny ładunek w stosunku do jonów determinujących potencjał. Zależy to od stosunku energii oddziaływania elektrostatycznego  i specyficzny potencjał adsorpcji
i specyficzny potencjał adsorpcji  . Jony gęstej warstwy są utrwalone. Pozostała część jonów znajduje się w warstwie rozproszonej, jony te są wolne i mogą przedostawać się w głąb roztworu, tj. z obszaru o wyższym stężeniu do obszaru o niższym stężeniu. Całkowita gęstość ładunku składa się z dwóch części.
. Jony gęstej warstwy są utrwalone. Pozostała część jonów znajduje się w warstwie rozproszonej, jony te są wolne i mogą przedostawać się w głąb roztworu, tj. z obszaru o wyższym stężeniu do obszaru o niższym stężeniu. Całkowita gęstość ładunku składa się z dwóch części.

 - Ładunek warstwy Helmholtza
- Ładunek warstwy Helmholtza
 -Ładunek warstwy rozproszonej
-Ładunek warstwy rozproszonej
Na powierzchni znajduje się pewna liczba centrów adsorpcji, z których każde oddziałuje z jednym przeciwjonem. Stała takiej reakcji quasi-chemicznej wynosi:

 , Gdzie
, Gdzie  - ułamek molowy przeciwjonów w roztworze
- ułamek molowy przeciwjonów w roztworze
Rozkład Helmholtza
Potencjał maleje liniowo
Dystrybucja potencjału Gouy’a. Nie ma gęstej warstwy, potencjał maleje wykładniczo od wartości 
Dystrybucja Sterna.
Początkowo potencjalny spadek ma charakter liniowy, a następnie wykładniczy.
Kiedy w przypadku elektroforezy przyłożone zostanie pole elektryczne, to nie cząstka fazy stałej porusza się bezpośrednio, ale cząstka fazy stałej wraz z warstwą otaczających ją jonów. DES powtarza kształt cząstki fazy rozproszonej. Po przyłożeniu potencjału część warstwy rozproszonej zostaje oderwana. Linia przerwania nazywa się przesuwająca się granica.
Potencjał powstający na granicy poślizgu w wyniku oderwania się części warstwy rozproszonej nazywa się potencjałem potencjał elektrokinetyczny(Potencjał zeta  ).
).
Cząstka fazy rozproszonej, otoczona warstwą przeciwjonów i podwójną warstwą elektryczną, nazywana jest cząstką fazy rozproszonej micela.
Zasady pisania miceli koloidalnych:


Elektrolit ładujący 1-1
T jest cząstką fazy rozproszonej.
AA jest granicą pomiędzy częściami gęstymi i rozproszonymi.
BB jest granicą poślizgu.
Granica poślizgu może, ale nie musi, pokrywać się z linią AA.
Wartość pH, przy której potencjał zeta wynosi zero, nazywa się punkt izoelektryczny.
CaCl2 + Na2SO4 → CaSO4 ↓ + 2NaCl
1. Powyżej CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n-x)Cl - ) 2 X + X Cl - - rekordowe micele.
CaSO 4 m - kruszywo.
Rdzeń stanowi CaSO 4 m∙nCa 2+.
CaSO 4 m∙nCa 2+ 2( n-x)Cl - cząstka.
2. Nadmiar Na2SO4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micela
CaSO 4 m - kruszywo.
CaSO 4 m∙nSO 4 2 + jest rdzeniem.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - cząstka
Równanie Helemholtza-Smoluchowskiego

 - prędkość liniowa przemieszczania się granic (w elektroosmozie).
- prędkość liniowa przemieszczania się granic (w elektroosmozie).
 - różnica potencjałów na płytkach kondensatora (w elektroosmozie).
- różnica potencjałów na płytkach kondensatora (w elektroosmozie).


 - objętościowe natężenie przepływu roztworu, S- kwadrat Przekrój komórki.
- objętościowe natężenie przepływu roztworu, S- kwadrat Przekrój komórki.

mi jest natężeniem pola elektrycznego.


 (na elektroosmozę).
(na elektroosmozę).
Dla potencjału przepływu:

 - potencjał
- potencjał
 - ciśnienie membranowe
- ciśnienie membranowe
Z reguły wartość ruchliwości elektroforetycznej i elektroosmotycznej jest mniejsza niż obliczona. Jest to spowodowane:
Efekt relaksacyjny (podczas ruchu cząstki fazy rozproszonej zostaje naruszona symetria atmosfery jonowej).
Hamowanie elektroforetyczne (pojawienie się dodatkowego tarcia w wyniku ruchu przeciwjonów).
Zakłócenia linii prądu w przypadku cząstek przewodzących prąd elektryczny.
Zależność napięcia powierzchniowego od potencjału. Równanie Lippmanna.
Tworzenie się DEL następuje spontanicznie w wyniku chęci układu do zmniejszenia swojej energii powierzchniowej. W kontekście stałości T I P uogólnione równanie pierwszej i drugiej zasady termodynamiki wygląda następująco:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1. równanie Lippmanna.
- 1. równanie Lippmanna.
 jest gęstością ładunku powierzchniowego.
jest gęstością ładunku powierzchniowego.
 - pojemność różnicowa.
- pojemność różnicowa.
 - 2. równanie Lippmanna.
- 2. równanie Lippmanna.
Z- pojemność.
Rozwiązujemy pierwsze równanie Lippmanna i podstawowe równanie adsorpcji:
 ,
,


 , Następnie
, Następnie
 - Równanie Nernsta
- Równanie Nernsta
 ,
,
 ,
,




 - równanie krzywej elektrokapilarnej (ECC).
- równanie krzywej elektrokapilarnej (ECC).
W  :
: , Ale
, Ale 
Kationowe środki powierzchniowo czynne (CSAS) redukują katodową gałąź ECC.
Anionowe środki powierzchniowo czynne (ASS) redukują anodową gałąź ECC.
Niejonowe środki powierzchniowo czynne (NSA) redukują środkową część ECC.
Stabilność układów rozproszonych. Ciśnienie klinujące.
Systemy rozproszone można podzielić:
Układy niestabilne termodynamicznie mogą być stabilne kinetycznie w wyniku przejścia do stanu metastabilnego.
Istnieją dwa rodzaje stabilności:
Stabilność sedymentacji (ze względu na grawitację).
Stabilność agregacyjna. (w odniesieniu do klejenia)
Koagulacja to proces sklejania się cząstek, prowadzący do utraty stabilności agregacji. Koagulacja może być spowodowana zmianami temperatury, pH, mieszaniem, ultradźwiękami.
Rozróżnij koagulację:
Odwracalny.
Nieodwracalny.
Koagulacja przebiega po wprowadzeniu elektrolitów.
Zasady krzepnięcia:

Film- jest to część systemu zlokalizowana pomiędzy dwoma interfejsami.
rozłączające ciśnienie występuje przy gwałtownym spadku grubości folii w wyniku interakcji zbliżających się warstw powierzchniowych.

«-» - gdy grubość folii maleje, ciśnienie rozłączające wzrasta.
P 0 to ciśnienie w fazie objętościowej, która jest kontynuacją międzywarstwy.
P 1 to ciśnienie w folii.
Teoria stabilności. DLFO (Deryagin, Landau, Fairway, Overbeck).
Zgodnie z teorią DLVO w ciśnieniu rozłączającym wyróżnia się dwie składowe:
elektrostatyczny PE (dodatni, wynika to z sił odpychania elektrostatycznego). Odpowiada spadkowi energii Gibbsa wraz ze wzrostem grubości warstwy.
Molekularny PM (ujemny, ze względu na działanie sił przyciągających). Jest to spowodowane ściskaniem folii pod wpływem chemicznych sił powierzchniowych, promień działania sił wynosi dziesiąte części nm przy energii rzędu 400 kJ/mol.
Całkowita energia interakcji:

 - system jest stabilny zagregowany
- system jest stabilny zagregowany
 - niestabilny system
- niestabilny system
P  składnik pozytywny.
składnik pozytywny.
Wzrost wynika ze wzrostu energii potencjalnej podczas ściskania cienkich folii. W przypadku grubych folii nadmiar energii jonów jest kompensowany i jest równy oddziaływaniu energii w większości ośrodka dyspersyjnego.
Jeśli  (
( - grubość folii,
- grubość folii,  - promień jonu) rozrzedzenie filmu prowadzi do zaniku i redukcji w nim cząsteczek i jonów przy minimalnej energii powierzchniowej. Zmniejsza się liczba sąsiadujących cząstek, w wyniku czego wzrasta energia potencjalna cząstek pozostających w folii.
- promień jonu) rozrzedzenie filmu prowadzi do zaniku i redukcji w nim cząsteczek i jonów przy minimalnej energii powierzchniowej. Zmniejsza się liczba sąsiadujących cząstek, w wyniku czego wzrasta energia potencjalna cząstek pozostających w folii.


Teoria DLVO traktuje oddziaływanie cząstek jako oddziaływanie płytek.

Cząsteczki nie oddziałują



 - równanie Laplace'a,
- równanie Laplace'a,  ,
,


Do słabo naładowanych powierzchni
W przypadku silnie naładowanych powierzchni:

Składnik molekularny to oddziaływanie dwóch atomów:
 ~
~
Oddziaływanie atomu z powierzchnią:

Weźmy dwa rekordy:
D  Aby otrzymać składnik molekularny, należy zsumować wszystkie energie interakcji atomów prawej i lewej płytki.
Aby otrzymać składnik molekularny, należy zsumować wszystkie energie interakcji atomów prawej i lewej płytki.
Gdzie  - Stała Hamakera (uwzględnia naturę oddziałujących ciał).
- Stała Hamakera (uwzględnia naturę oddziałujących ciał).
To. energię interakcji cząstek w układzie można wyrazić za pomocą krzywych potencjału.
I jest pierwotnym minimum potencjału. Jest to strefa nieodwracalnej koagulacji, w której przeważają siły przyciągania.
II - strefa stabilności agregatowej, przeważają siły odpychające.
III - minimum potencjału wtórnego (lub strefa flokulacji). Pomiędzy cząstkami fazy rozproszonej znajduje się warstwa elektrolitu, przy czym cząstki mogą zostać oddzielone i przeniesione do strefy stabilności agregacyjnej.

Krzywa 1 – system jest agregatywnie stabilny.
Krzywa 2 jest stabilna w strefie I, niestabilna w strefie II.
Krzywa 3 – w układzie nastąpiła koagulacja.
Krzywa 4 – w punkcie 4 całkowita energia oddziaływania U=0,  , ten ekstremalny punkt odpowiada początkowi szybkiej koagulacji.
, ten ekstremalny punkt odpowiada początkowi szybkiej koagulacji.
Istnieją dwa przypadki:
1. Powierzchnie są słabo naładowane:
U \u003d U E + U M \u003d 0
 (1)
(1)
2)

 (2)
(2)



 - jest to grubość warstwy odpowiadająca początkowi procesu koagulacji.
- jest to grubość warstwy odpowiadająca początkowi procesu koagulacji.






 - do słabo naładowanych powierzchni
- do słabo naładowanych powierzchni
 Następnie
Następnie 
2. W przypadku silnie naładowanych powierzchni:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Ustawmy kwadrat (3)

Koagulacja:
W przypadku adsorpcji specyficznej jony mogą zostać zaadsorbowane w ilości nadrównoważnej w taki sposób, że powierzchnia może zmienić swój ładunek. Trwa ładowanie powierzchni.
W przypadku adsorpcji specyficznej adsorbowane mogą być nie tylko jony o przeciwnych znakach, ale także jony jednego jonu.
Jeśli zaadsorbowane zostaną jony o tym samym znaku co powierzchnia, to w warstwie powierzchniowej nie nastąpi spadek potencjału, ale jego wzrost.

Koagulacja neutralizująca (zachodzi przy udziale cząstek słabo naładowanych i zależy nie tylko od ładunku elektrolitu koagulującego, ale także od potencjału na granicy warstw gęstej i rozproszonej).
Teoria szybkiej koagulacji Smoluchowskiego.

Zależność szybkości krzepnięcia od stężenia elektrolitu.
I – szybkość krzepnięcia jest niska,
II - szybkość koagulacji jest praktycznie proporcjonalna do stężenia elektrolitu.
III - obszar szybkiej koagulacji, szybkość jest praktycznie niezależna od stężenia.
Podstawowe postanowienia:
Początkowy zol jest monodyspersyjny, podobne cząstki mają kształt kulisty.
Wszystkie zderzenia cząstek są skuteczne.
Kiedy zderzają się dwie cząstki pierwotne, powstaje cząstka wtórna. Szkoła średnia + podstawowa = trzeciorzędna. Pierwotny, wtórny, trzeciorzędny - wielość.
Pod względem kinetyki chemicznej proces koagulacji można opisać równaniem:
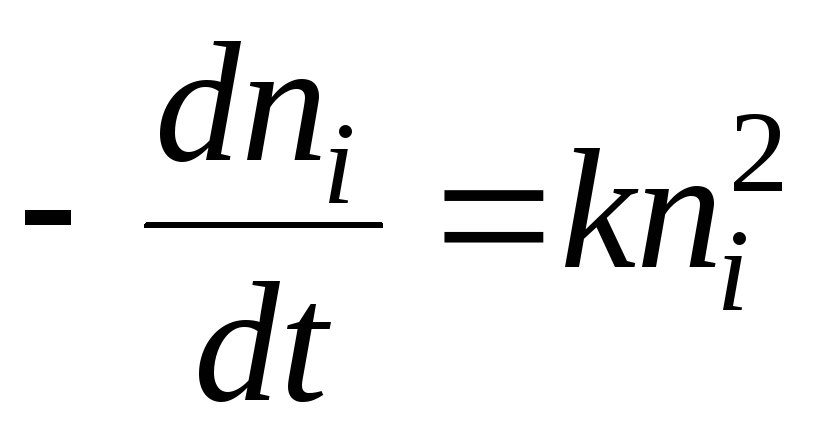
Rozwiązaniem będzie równanie: 
 - czas półkoagulacji. Jest to czas, w którym liczba cząstek zolu zmniejsza się 2-krotnie.
- czas półkoagulacji. Jest to czas, w którym liczba cząstek zolu zmniejsza się 2-krotnie.

 ,
,
 ,
,
 ,
,


Wraz ze wzrostem krotności maksimum krzywych koagulacji przesuwa się w stronę większych wartości  .
.
Wady:
Założenie monodyspersyjności.
Założenie o skuteczności wszystkich zderzeń.